Perdita di funzione e fenotipo clinico
|
|
|
- Leonzia Fiorini
- 8 anni fa
- Visualizzazioni
Transcript
1 Disordini genomici un disordine genomico submicroscopico è una patologia causata da Perdita Acquisizione Alterazione di uno o più geni contigui, le cui variazioni di dosaggio possono produrre effetti fenotipici. La base molecolare è rappresentata da riarrangiamenti genomici, quali delezioni,duplicazioni, inversioni, spesso senza alterazioni apparenti del cariotipo (<5Mb).
.")
2 Perdita di funzione e fenotipo clinico La gran parte dei geni autosomici si trova nella condizione A o C: il dosaggio genico critico è <50%. In tal caso, si osserva un fenotipo patologico solo se entrambi gli alleli sono colpiti (recesssivo). I geni autosomici responsabili della patogenesi dei disordini genomici si trovano nella condizione B o D: si osserva un fenotipo già in eterozigosi per aploinsufficienza. Spesso anche un dosaggio genico aumentato >>100% può determinare una patologia.
3 Aploinsufficienza Insufficiente quantità di prodotto genico causata da una mutazione in eterozigosi. La mutazione è di tipo allele amorfo o ipomorfo. Colpisce geni per i quali il 50% di prodotto genico non è sufficiente a garantirne la funzione Un dosaggio preciso è richiesto ai fattori di trascrizione e alle molecole di segnale espressi nel corso dello sviluppo.
.")
4 Delezioni del cromosoma X: nei maschi si osserva direttamente in fenotipo come sindrome da geni contigui. Delezioni autosomiche in eterozigosi: Molto spesso il dosaggio dimezzato non è causa di malattia (CNV). In una sindrome da delezione, è risolutivo trovare la stessa sindrome causata da una mutazione puntiforme in uno solo dei geni (S. di Alagille: microdelezione 20p11, mutazioni JAG1). Se questa non si trova, la sindrome esiste solo come somma di più difetti.
5 Duplicazioni segmentali (LCRs) il genoma umano contiene complessivamente il 13,7% di segmenti duplicati più del 90-95% di identità di sequenza il 5,2% del genoma contiene segmenti duplicati lunghi tra 1 e 10kb, mentre il 4,5% tra 10kb e 20kb i cromosomi più colpiti sono l Y (50,4%) ed il 22 (11,9%), ma anche il 7, 9, 10, 15, 16, 17 e X le duplicazioni segmentali possono essere intracromosomiche o intercromosomiche con tre localizzazioni differenti: pericentromeriche (47Mb, dupliconi originati da altri cromosomi) subtelomeriche (ciascuna solo kb, orientate) interstiziali (solo nella specie umana sono disseminate ad una distanza media di 3Mb)
6 Non-allelic homologous recombination (NAHR)
7 Non-allelic homologous recombination (NAHR)

8 Riarrangiamenti cromosomici mediati da duplicazioni segmentali

9 9
10 Principali sindromi da microdelezione LOCALIZZAZIONE SOGGETTI CON SINDROME CROMOSOMICA MICRODELEZIONE Prader Willi/Angelman 15q % Williams 7q % DiGeorge/Velocardiofacciale 22q % Smith-Magenis 17p %
 E la più frequente sindrome da microdelezione (1:4000) La")

11 Di George/Velocardiofacciale (del22q11.2) E la più frequente sindrome da microdelezione (1:4000) La delezione comprende 3Mb ed almeno 30 geni Nella maggior parte dei casi de novo. In circa il 10% ereditata da un genitore moderatamente affetto.
12 del22q11.2 Il fenotipo clinico varia da lieve a grave Cardiopatie (77% dei casi, soprattutto difetti troncoconali) tronco arterioso, tetralogia di Fallot, difetto del setto ventricolare. Anomalie del palato (75% dei casi) palatoschisi aperta, labiopalatoschisi, insufficienza velofaringea. Ritardo dello sviluppo. Dismorfismi facciali (ipertelorismo, epicanto, radice nasale prominente, ecc.) Anomalie vertebrali (vertebre a farfalla, emivertebre) Deficit immunitario (75% dei casi) da aplasia/ipoplasia del timo Ipocalcemia (50% dei casi) alla nascita. Altri segni clinici: anomalie gastrointestinali, sordità, anomalie renali (agenesia renale), anomalie dei denti (ipoplasia dello smalto) Disturbi dell'apprendimento e/o disturbi psichiatrici (disturbo deficit dell'attenzioneiperattività, schizofrenia) La diagnosi si basa sull'esame clinico e sulla presenza di alterazioni cardiache rilevabili con l'ecocardiografia, o anomalie vertebrali osservabili con la radiografia della regione cervicale. È confermata dall'identificazione della delezione 22q


13 Mutazioni in TBX1 causano 5 caratteristiche fenotipiche principali: facies anomala difetti cardiaci ipoplasia del timo insufficienza velofaringea con palatoschisi disfunzione paratiroidea con ipocalcemia. 13
14 Embrione a 4-6 settimane di gestazione Alterazioni della migrazione delle cellule della cresta neurale dal romboencefalo al sistema degli archi faringei, probabilmente concorrono a determinare peculiari elementi del fenotipo: Anomalie cardiache Anomalie del palato e dismorfismi facciali Deficit immunitario e ipocalcemia.
15 Williams-Beuren La sindrome di Williams è un disordine multisistemico dello sviluppo con un incidenza di circa 1/ nati. E causata da una microdelezione di Mb a 7q11.23, che coinvolge geni. Principali caratteristiche cliniche: Facies caratteristica (faccia da Elfo) Anomalie cardiovascolari Stenosi sopravalvolare dell aorta (70% dei pazienti) Stenosi periferica delle arterie polmonari Ipertensione (50% dei pazienti) Anomalie endocrine Episodi di ipercalcemia (5-50% dei pazienti, particolarmente nell infanzia) Ridotta tolleranza al glucosio isolitamente aumentata nei pazienti WS Anomalie neurocognitive

16 Williams-Beuren Syndrome Critical Region ELN (gene dell elastina): mutazioni in pazienti con stenosi sopravalvolare aortica familiare. LIMK1 e CLIP2: riduzione delle abilità motorie e spaziovisive in modelli animali. GTF2I (GTF2IRD1 incluso): anomalie craniofacciali, disabilità intellettiva, profilo cognitivo tipico WBS (modelli animali e delezioni atipiche).
17 Diagnosi molecolare La microdelezione non è rilevabile con l analisi del cariotipo
18 Caratteristiche morfologiche Faccia da Elfo Occhi blu (77%) con pattern stellato dell iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa bocca larga e guance piene microdontia e micrognazia Statura 10 cm in meno del normale
19 Aspetti cognitivi e comportamentali Ritardo mentale di grado lieve-moderato (IQ tra 41 e 80) Scarsa capacità di concentrazione Ritardo nell apprendimento del linguaggio,e poi esagerata loquacità Personalità amichevole e affettuosa: danno facilmente confidenza anche a sconosciuti Ansietà: spesso preoccupati per il benessere altrui Ipersensibilità ai suoni Memoria visiva e uditiva, spesso fuori dal comune Ricordano persone, luoghi e motivi musicali Predisposizione ad imparare le lingue e la musica
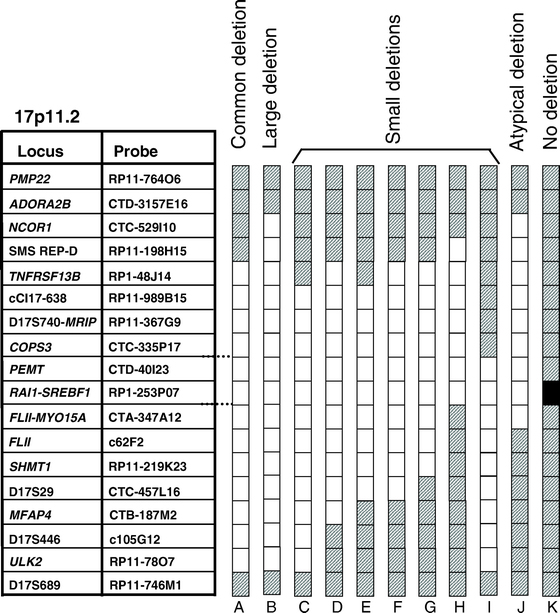
20 Smith-Magenis (SMS) E un disordine caratterizzato da anomalie multiple congenite e ritardo mentale E causato da microdelezione a 17p11.2 (90% dei casi) o mutazioni nel gene RAI1 (10% dei casi) con aploinsufficienza Disturbo sporadico (de novo) con una prevalenza di 1: Caratteristiche dei pazienti SMS: Ritardo mentale lieve-moderato (IQ 20-78) Caratteristiche comportamentali peculiari Anomalie cranio-facciali e scheletriche Ritardo nello sviluppo del linguaggio Disturbi del sonno (inversione nel ritmo circadiano della melatonina). Riduzione/perdita dell udito e infezioni croniche dell'orecchio Ipotonia Altre alterazioni sistemiche meno frequenti (es. oculari, cardiache, renali)

21 Circa il 90% dei pazienti SMS hanno la microdelezione 17p11.2 (tra 1.5 e 9 Mb), mentre il restante 10% ha mutazioni in RAI1 (Retinoic Acid Inducible 1). Circa il 70% dei pazienti la microdelezione è di circa 3.7Mb, mentre il restante 30% può presentare delezioni più grandi, più piccole o atipiche. Diagnosi molecolare mediante FISH, array CGH, MLPA.
22 22
23 Aspetti comportamentali I problemi comportamentali sono una caratteristica dei pazienti SMS. Comportamenti disadattivi: Frequenti scoppi di collera, Ricerca di attenzione Aggressività, disobbedienza e scarsa attenzione Comportamenti autolesionistici (sbattere la testa, strappare la pelle, mordersi il polso) sono presenti già a mesi. Caratteristiche uniche della SMS: Onicotillomania (strapparsi le unghie delle mani e dei piedi) Poliembolocoilamania (inserimento di oggetti in orifizi del corpo) Abbracciarsi e stringere le mani
24 Sindrome 5p- Sindrome del Cri du Chat Frequenza 1/50000 nati vivi. E espressione di una delezione parziale del braccio corto del cromosoma 5, de novo nella maggioranza dei casi (85%). Può essere dovuta a: Segregazione sbilanciata di un riarrangiamento cromosomico presente in uno dei due genitori (5-12%). Riarrangiamenti strutturali de novo (6%). Formazione di cromosomi ad anello (2,4 %) Un 4% dei soggetti evidenzia mosaicismo. Il fenotipo, sebbene caratteristico evidenzia ampia variabilità. Superata l età neonatale la sopravvivenza non è compromessa. 24
25 Sindrome 5p- o Sindrome del Cri du Chat (2) I neonati hanno un pianto acuto e flebile, che è caratteristico di questa sindrome. Grave ritardo di crescita e nello sviluppo psicomotorio. Microcefalia Faccia tondeggiante con ipertelorismo, radice del naso allargata, strabismo divergente, micrognazia, ipotonia congenita. Manifestazioni cliniche tardive sono: Asimmetria facciale, voce flebile e acuta, scoliosi. Grave deficit del linguaggio e deficit intellettivo medio-grave. La diagnosi clinica è validata dall analisi citogenetica per evidenziare e definire l estensione della delezione. Regione critica per la sindrome del cri du chat è la banda 5p
26 Sindrome da monosomia 4p o Sindrome di Wolf-Hirshhorn Frequenza 1/50000 nati. La WHS è causata da una delezione parziale del braccio corto del cromosoma 4 che può avere estensione variabile. La regione critica per la tipica sintomatologia e identificabile in 4p16.3. La maggioranza delle delezioni insorge de novo (90%). Più raramente è il risultato della segregazione sbilanciata di una traslocazione familiare. Rara la descrizione di cromosomi ad anello. Quadro clinico variabile in funzione dell estensione della delezione. Ritardo di crescita intrauterino (IUGR) e scarso accrescimento dopo la nascita. Ritardo mentale Microcefalia con dolicocefalia a frequente asimmetria cranica. Dismorfismo facciale (fronte ampia, ipertelorismo, globi oculari sporgenti). 26
27 27
28 Diagnostica molecolare dei disordini genomici
29 MLPA (Multiplex Ligation-dependent Probe Amplification) Riconoscimento di variazioni del numero di copie in più di 40 distinte sequenze genomiche mediante un unica reazione di PCR. Campi applicativi: Caratterizzazione di delezioni/duplicazioni o variazioni del numero di copie (aneuploidie) Determinare lo stato di metilazione di promotori o regioni imprinted Riconoscimento di specifiche mutazioni puntiformi o SNPs (single nucleotide polymorphism) Richiede minime quantità di DNA genomico (circa 20 ng).

30 Come funziona l MLPA

31 MLPA probes
32 Hybridization 1. The MLPA probemix is added to denatured genomic DNA 2. The two parts of each probe hybridize to adjacent target sequences

33 Ligation 3. Probes are ligated by a thermostable ligase

34 PCR amplification 4. A universal primer pair is used to amplify all ligated probes The PCR product of each probe has a unique length (rangeb bp)
35 Separation and quantification by capillary electrophoresis Each peak is the amplification product of a specific probe. Samples are compared to a control sample. A difference in relative peak height or peak area indicates a copy number change of the probe target sequence
36 36
37 37
38 Analisi mediante MLPA del gene DMD (1) 1.6 ìmlpa_p34_3_c^gmstxt.txt Mix P Paziente A P35 Mix P Ratio Ratio X DMD exon 01 X DMD exon 03 X DMD exon 05 X DMD exon 07 X DMD exon 09 X DMD exon 21 X DMD exon 23 X DMD exon 25 X DMD exon 27 X DMD exon 29 X DMD exon 41 X DMD exon 43 X DMD exon 45 X DMD exon 47 X DMD exon 49 X DMD exon 61 X DMD exon 63 X DMD exon 65 X DMD exon 67 X DMD exon 69 c c c 0 X DMD exon 11 X DMD exon 13 X DMD exon 15 X DMD exon 17 X DMD exon 19 X DMD exon 31 X DMD exon 33 X DMD exon 35 X DMD exon 37 X DMD exon 39 X DMD exon 51 X DMD exon 53 X DMD exon 55 X DMD exon 57 X DMD exon 59 X DMD exon 71 X DMD exon 73 X DMD exon 75 X DMD exon 77 X DMD exon 79 c c c Mapview Mapview Femmina portatrice di una delezione degli esoni 12-30
39 Analisi mediante MLPA del gene DMD (2) Maschio affetto (BMD) con duplicazione in emizigosi degli esoni 3-4
40 Methylation-specific MLPA (MS-MLPA)
41 Methylation-specific MLPA (MS-MLPA) Promoter region of SNRPN gene Undigested or Digested with HhaI (CGC G) AS PWS Normal
42 Identificazione di specifiche mutazioni puntiformi mediante MLPA
43
Diagnostica molecolare dei disordini genomici
 Diagnostica molecolare dei disordini genomici MLPA (Multiplex Ligation-dependent Probe Amplification) Riconoscimento di variazioni del numero di copie in più di 40 distinte sequenze genomiche mediante
Diagnostica molecolare dei disordini genomici MLPA (Multiplex Ligation-dependent Probe Amplification) Riconoscimento di variazioni del numero di copie in più di 40 distinte sequenze genomiche mediante
PATOLOGIA CROMOSOMICA
 PATOLOGIA CROMOSOMICA CARIOTIPO UMANO NORMALE 4 6,X Y 4 6,X X CARIOTIPO UMANO NORMALE Cariotipo normale, giemsa CARIOTIPO UMANO NORMALE bande G C r o m o s o m a c o n b and e G t e lo m e r o c e ntr
PATOLOGIA CROMOSOMICA CARIOTIPO UMANO NORMALE 4 6,X Y 4 6,X X CARIOTIPO UMANO NORMALE Cariotipo normale, giemsa CARIOTIPO UMANO NORMALE bande G C r o m o s o m a c o n b and e G t e lo m e r o c e ntr
5 modulo didattico - Patologia cromosomica.
 5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
SCAMBI tra due coppie di cromosomi NON omologhi
 TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide
 Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
- Markers di anomalie cromosomiche. - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards)
 - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards)") - Markers di anomalie cromosomiche - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards) - Sindrome di Turner - Sindrome di Williams - Sindrome di Angelman
- Markers di anomalie cromosomiche - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards) - Sindrome di Turner - Sindrome di Williams - Sindrome di Angelman
Delezione. Duplicazione. Variazioni della struttura. Inversione. Traslocazione. Mutazioni cromosomiche. Nullisomia Monosomia Trisomia Tetrasomia
 Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale. Dalla citogenetica convenzionale alla citogenetica molecolare!
 e diagnosi prenatale. Dalla citogenetica convenzionale alla citogenetica molecolare!") Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale Dalla citogenetica convenzionale alla citogenetica molecolare! CARIOTIPO STANDARD Identifica anomalie strutturali bilanciate e sbilanciate
Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale Dalla citogenetica convenzionale alla citogenetica molecolare! CARIOTIPO STANDARD Identifica anomalie strutturali bilanciate e sbilanciate
INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI
 SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI") INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene eseguita su cellule
INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene eseguita su cellule
4 modulo didattico - Modalità di trasmissione delle malattie
 4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
Mutazioni cromosomiche. Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo
 Mutazioni cromosomiche Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo Mutazioni cromosomiche Variazioni della struttura Variazioni
Mutazioni cromosomiche Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo Mutazioni cromosomiche Variazioni della struttura Variazioni
Anomalie cromosomiche
 Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI
 VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI Le alterazioni strutturali implicano cambiamenti di parti di cromosomi. Esistono 4 tipi di tali mutazioni: Delezione Duplicazione inversione Traslocazione Determinano
VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI Le alterazioni strutturali implicano cambiamenti di parti di cromosomi. Esistono 4 tipi di tali mutazioni: Delezione Duplicazione inversione Traslocazione Determinano
PerCorso Endocrinologia Pediatrica: Caso Clinico. Deficit di accrescimento
 PerCorso Endocrinologia Pediatrica: Caso Clinico Deficit di accrescimento Verona Rossella.Gaudino@univr.it Caso Clinico: MARIA Maria è una bambina di 7 anni che giunge alla nostra osservazione per persistenza
PerCorso Endocrinologia Pediatrica: Caso Clinico Deficit di accrescimento Verona Rossella.Gaudino@univr.it Caso Clinico: MARIA Maria è una bambina di 7 anni che giunge alla nostra osservazione per persistenza
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico. Volume sulle Disabilità intellettive, cap. 5.
 Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
FINALITA : evidenziare la. cromosomiche fetali.
 Il Cariotipo Fetale Molecolare l (array-cgh) Il Cariotipo Fetale e TRADIZIONALE FINALITA : evidenziare la presenza di eventuali anomalie cromosomiche fetali. TECNICA: comporta la coltura delle cellule
Il Cariotipo Fetale Molecolare l (array-cgh) Il Cariotipo Fetale e TRADIZIONALE FINALITA : evidenziare la presenza di eventuali anomalie cromosomiche fetali. TECNICA: comporta la coltura delle cellule
ll consumo di bevande alcoliche durante la gravidanza e l allattamento può avere effetti dannosi sulla salute del bambino.
 NON BERE BEVANDE ALCOLICHE IN GRAVIDANZA ED IN ALLATTAMENTO U.O. Consultorio Familiare Distretto n 1 e n 2 Ostetrica t CRISTINA FILIPPI Ostetrica BIANCA PIVA 1 Non bere bevande alcoliche in gravidanza
NON BERE BEVANDE ALCOLICHE IN GRAVIDANZA ED IN ALLATTAMENTO U.O. Consultorio Familiare Distretto n 1 e n 2 Ostetrica t CRISTINA FILIPPI Ostetrica BIANCA PIVA 1 Non bere bevande alcoliche in gravidanza
22q11.2 MICRODELETIONS AND MILD HYPOTHYROIDISM IN PEDIATRIC PATIENTS WITH CONGENITAL HEART DEFECTS
 22q11.2 MICRODELETIONS AND MILD HYPOTHYROIDISM IN PEDIATRIC PATIENTS WITH CONGENITAL HEART DEFECTS Marcello Frigerio Laboratorio di Biologia Molecolare IRCCS Policlinico San Donato marcello.frigerio@gmail.com
22q11.2 MICRODELETIONS AND MILD HYPOTHYROIDISM IN PEDIATRIC PATIENTS WITH CONGENITAL HEART DEFECTS Marcello Frigerio Laboratorio di Biologia Molecolare IRCCS Policlinico San Donato marcello.frigerio@gmail.com
Anomalie cromosomiche
 Anomalie cromosomiche Alterazioni che interessano il DNA genomico determinando la perdita o l acquisizione di interi cromosomi o segmenti di essi. Se l alterazione è tale da poter essere visibile al microscopio
Anomalie cromosomiche Alterazioni che interessano il DNA genomico determinando la perdita o l acquisizione di interi cromosomi o segmenti di essi. Se l alterazione è tale da poter essere visibile al microscopio
GENETICA CITOGENETICA MEDICA
 CORSO INTEGRATO DI GENETICA E BIOLOGIA MOLECOLARE GENETICA A.A.2015/2016 Prof Alberto E.Turco Me 21.10.2015 Lezioni 15 e 16 CITOGENETICA MEDICA ANOMALIE CROMOSOMICHE (Chromosome Disorders) - Abortività
CORSO INTEGRATO DI GENETICA E BIOLOGIA MOLECOLARE GENETICA A.A.2015/2016 Prof Alberto E.Turco Me 21.10.2015 Lezioni 15 e 16 CITOGENETICA MEDICA ANOMALIE CROMOSOMICHE (Chromosome Disorders) - Abortività
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI
 DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
Sindrome di Down. Aspetti genetici, fisici, motori e medici. www.fluitekruidje.com
 Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Genetica Medica corsi di laurea triennali
 Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Aggiornamenti in ambito genetico
 10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
eterogeneità genetica
 eterogeneità genetica Fenotipo clinico indistinguibile con pattern di trasmissione ereditaria differente: autosomico dominante, autosomico recessivo, X-linked, o mitocondriale Esempio: le atassie cerebellari
eterogeneità genetica Fenotipo clinico indistinguibile con pattern di trasmissione ereditaria differente: autosomico dominante, autosomico recessivo, X-linked, o mitocondriale Esempio: le atassie cerebellari
Breve panoramica sulle categorie diagnostiche attuali dei DPS. Definizione e descrizione dei criteri diagnostici maggiormente applicati
 Breve panoramica sulle categorie diagnostiche attuali dei DPS Definizione e descrizione dei criteri diagnostici maggiormente applicati Sistemi Internazionali di classificazione dei disturbi mentali Il
Breve panoramica sulle categorie diagnostiche attuali dei DPS Definizione e descrizione dei criteri diagnostici maggiormente applicati Sistemi Internazionali di classificazione dei disturbi mentali Il
-riesce a identificare riarrangiamenti cromosomici sbilanciati, ovvero delezioni o duplicazioni
 L array-cgh: -riesce a identificare riarrangiamenti cromosomici sbilanciati, ovvero delezioni o duplicazioni -ha un potere si risoluzione 100 volte superiore al cariotipo (100 Kb versus 10 Mb) Microarray-based
L array-cgh: -riesce a identificare riarrangiamenti cromosomici sbilanciati, ovvero delezioni o duplicazioni -ha un potere si risoluzione 100 volte superiore al cariotipo (100 Kb versus 10 Mb) Microarray-based
LE MALATTIE GENETICHE CLASSE 3 C
 LE MALATTIE GENETICHE CLASSE 3 C Malattia causata da allele dominante Il nanismo acondroplastico è una malattia causata da un allele dominante; gli individui che ne sono affetti sono di statura molto bassa,
LE MALATTIE GENETICHE CLASSE 3 C Malattia causata da allele dominante Il nanismo acondroplastico è una malattia causata da un allele dominante; gli individui che ne sono affetti sono di statura molto bassa,
A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo
 2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel
 Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
GENETICA MENDELIANA NELL UOMO
 GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali. Silvia Sansavini
 CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
= femmina. = maschio. = fenotipo banda bianca. = fenotipo pezzato. =fenotipo colore uniforme
 Test n.8 Dalle Olimpiadi delle Scienze Naturali 2002 PARTE TERZA Le 5 domande di questa parte riguardano il medesimo argomento e sono introdotte da un breve testo e da uno schema. In una razza bovina il
Test n.8 Dalle Olimpiadi delle Scienze Naturali 2002 PARTE TERZA Le 5 domande di questa parte riguardano il medesimo argomento e sono introdotte da un breve testo e da uno schema. In una razza bovina il
Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
 Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked") Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Patologia dell'asse ipotalamoipofisi-igf-cartilagine. Luca Taf Azienda USL 8 Arezzo UO Pediatria
 Patologia dell'asse ipotalamoipofisi-igf-cartilagine Luca Taf Azienda USL 8 Arezzo UO Pediatria Sistema complesso Azione del GH a livello cellulare Alcuni geni coinvolti nel processo di crescita GH-1 GHRH
Patologia dell'asse ipotalamoipofisi-igf-cartilagine Luca Taf Azienda USL 8 Arezzo UO Pediatria Sistema complesso Azione del GH a livello cellulare Alcuni geni coinvolti nel processo di crescita GH-1 GHRH
Gene Mutations and Disease
 Gene Mutations and Disease Mutazioni somatiche: Nuove mutazioni che insorgono casualmente nelle cellule somatiche o nella linea germinale di singoli individui. Le mutazioni germinali possono essere trasmesse
Gene Mutations and Disease Mutazioni somatiche: Nuove mutazioni che insorgono casualmente nelle cellule somatiche o nella linea germinale di singoli individui. Le mutazioni germinali possono essere trasmesse
Seconda Parte Specifica di scuola - Genetica medica - 31/07/2015
 Domande relative alla specializzazione in: Genetica medica Domanda #1 (codice domanda: n.311) : Qualora una donna risultasse positiva al test genetico per la ricerca di mutazioni del gene BRCA1, per quale
Domande relative alla specializzazione in: Genetica medica Domanda #1 (codice domanda: n.311) : Qualora una donna risultasse positiva al test genetico per la ricerca di mutazioni del gene BRCA1, per quale
Mutation screening mediante PCR
 Mutation screening mediante PCR Denaturing High-Perfomance Liquid Chromatography (DHPLC) E una tecnica di analisi cromatografica ad alta pressione che consente di discriminare tra homoduplex ed heteroduplex
Mutation screening mediante PCR Denaturing High-Perfomance Liquid Chromatography (DHPLC) E una tecnica di analisi cromatografica ad alta pressione che consente di discriminare tra homoduplex ed heteroduplex
Il feto con una singola anomalia correggibile: problematiche del consiglio prenatale
 Il feto con una singola anomalia correggibile: problematiche del consiglio prenatale Dr.ssa Maria Segata Centri Medici GynePro, Bologna e Università di Bologna Diagnosi prenatale Anomalia Ecografia esperta
Il feto con una singola anomalia correggibile: problematiche del consiglio prenatale Dr.ssa Maria Segata Centri Medici GynePro, Bologna e Università di Bologna Diagnosi prenatale Anomalia Ecografia esperta
Titolo progetto: Correlazione genotipo-fenotipo in pazienti con la Sindrome di PraderWilli
 Titolo progetto: Correlazione genotipo-fenotipo in pazienti con la Sindrome di PraderWilli Descrizione: La Sindrome di Prader-Willi (PW) è una malattia genetica, colpisce entrambi i sessi ed ha un incidenza
Titolo progetto: Correlazione genotipo-fenotipo in pazienti con la Sindrome di PraderWilli Descrizione: La Sindrome di Prader-Willi (PW) è una malattia genetica, colpisce entrambi i sessi ed ha un incidenza
Progetto sulle esostosi multiple
 Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
Alice Laita Raffaella Fornaseri Sofia Raffa
 PSICOPATOLOGICO PSICOPATOLOGICO Alice Laita Raffaella Fornaseri Sofia Raffa CAPITOLO V CAPITOLO V RISCHIO RISCHIO SINDROME DI DOWN È una malattia genetica causata dalla presenza di un cromosoma 21 in più:
PSICOPATOLOGICO PSICOPATOLOGICO Alice Laita Raffaella Fornaseri Sofia Raffa CAPITOLO V CAPITOLO V RISCHIO RISCHIO SINDROME DI DOWN È una malattia genetica causata dalla presenza di un cromosoma 21 in più:
LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2
 LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2 PRIMA INFANZIA: LESIONI CUTANEE DALLO SVILUPPO CARATTERISTICO ( dermatite bollosa congenita )
LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2 PRIMA INFANZIA: LESIONI CUTANEE DALLO SVILUPPO CARATTERISTICO ( dermatite bollosa congenita )
Tecniche di bandeggio
 Tecniche di bandeggio sono sistemi di colorazione che conferiscono ai cromosomi caratteristici pattern di bande più o meno intense ogni cromosoma umano presenta un bandeggio (ossia una sequenza di bande)
Tecniche di bandeggio sono sistemi di colorazione che conferiscono ai cromosomi caratteristici pattern di bande più o meno intense ogni cromosoma umano presenta un bandeggio (ossia una sequenza di bande)
La mutazione è una modificazione della sequenza delle basi del DNA
 La mutazione è una modificazione della sequenza delle basi del DNA Le mutazioni sono eventi rari e importanti in quanto sono alla base dell evoluzione biologica Le mutazioni possono essere spontanee (dovute
La mutazione è una modificazione della sequenza delle basi del DNA Le mutazioni sono eventi rari e importanti in quanto sono alla base dell evoluzione biologica Le mutazioni possono essere spontanee (dovute
LA SINDROME DI DOWN LA STORIA
 LA SINDROME DI DOWN LA STORIA La sindrome di Down, che è detta anche trisomia 21 o mongoloidismo, è una malattia causata dalla presenza di una terza copia del cromosoma 21; è la più comune anomalia cromosomica
LA SINDROME DI DOWN LA STORIA La sindrome di Down, che è detta anche trisomia 21 o mongoloidismo, è una malattia causata dalla presenza di una terza copia del cromosoma 21; è la più comune anomalia cromosomica
Alcol, gravidanza e allattamento
 Alcol 0 Giornata di studio 1 aprile 2015 Azienda Ospedaliero-Universitaria careggi-firenze Dr Maurizio Fontanarosa - Maternità di Careggi Alcol, gravidanza e allattamento Spettro dei disordini feto-alcolici
Alcol 0 Giornata di studio 1 aprile 2015 Azienda Ospedaliero-Universitaria careggi-firenze Dr Maurizio Fontanarosa - Maternità di Careggi Alcol, gravidanza e allattamento Spettro dei disordini feto-alcolici
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali
 CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
DIFETTI VISIVI NEI BAMBINI AFFETTI DALLA SINDROME DOWN
 DIFETTI VISIVI NEI BAMBINI AFFETTI DALLA SINDROME DOWN DADDY FADEL OPTOMETRISTA Nel 1866 John Langdon Down ha descritto per la prima volta la sindrome che porta il suo nome. Nel 1959 Lejeune et al. hanno
DIFETTI VISIVI NEI BAMBINI AFFETTI DALLA SINDROME DOWN DADDY FADEL OPTOMETRISTA Nel 1866 John Langdon Down ha descritto per la prima volta la sindrome che porta il suo nome. Nel 1959 Lejeune et al. hanno
Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche
 Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche La patogenesi di aborti dovuti ad anomalie cromosomiche è dovuta a: A) Ipoplasia placentare; B) Malformazioni
Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche La patogenesi di aborti dovuti ad anomalie cromosomiche è dovuta a: A) Ipoplasia placentare; B) Malformazioni
La genetica della Sindrome di Prader-Willi
 La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa
 Epigenetica ed espressione genica monoallelica Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa monoallelicamente,
Epigenetica ed espressione genica monoallelica Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa monoallelicamente,
Il vostro bambino e lo Screening Neonatale
 Il vostro bambino e lo Screening Neonatale Guida per i Genitori A cura di: Centro Fibrosi Cistica e Centro Malattie Metaboliche AOU A. Meyer, Firenze Cari genitori, la Regione Toscana, secondo un programma
Il vostro bambino e lo Screening Neonatale Guida per i Genitori A cura di: Centro Fibrosi Cistica e Centro Malattie Metaboliche AOU A. Meyer, Firenze Cari genitori, la Regione Toscana, secondo un programma
LA SINDROME DI VON HIPPEL-LINDAU. Conoscere per Curare
 LA SINDROME DI VON HIPPEL-LINDAU Conoscere per Curare 1) Che cos è VHL? La sindrome di Von Hippel-Lindau è una rara malattia a carattere ereditario che determina una predisposizione allo sviluppo di neoplasie
LA SINDROME DI VON HIPPEL-LINDAU Conoscere per Curare 1) Che cos è VHL? La sindrome di Von Hippel-Lindau è una rara malattia a carattere ereditario che determina una predisposizione allo sviluppo di neoplasie
Caso clinico traslocazione criptica t (1;10) U.O. Neonatologia TIN Azienda Ospedaliera BologninI Seriate (BG) Direttore Dott.ssa A.
 U.O. Neonatologia TIN Azienda Ospedaliera BologninI Seriate (BG) Direttore Dott.ssa A.") Caso clinico traslocazione criptica t (1;10) U.O. Neonatologia TIN Azienda Ospedaliera BologninI Seriate (BG) Direttore Dott.ssa A. Auriemma INTRODUZIONE La monosomia distale del braccio corto del cromosoma
Caso clinico traslocazione criptica t (1;10) U.O. Neonatologia TIN Azienda Ospedaliera BologninI Seriate (BG) Direttore Dott.ssa A. Auriemma INTRODUZIONE La monosomia distale del braccio corto del cromosoma
Alberto Viale I CROMOSOMI
 Alberto Viale I CROMOSOMI DA MENDEL ALLA GENETICA AL DNA ALLE MUTAZIONI I cromosomi sono dei particolari bastoncelli colorati situati nel nucleo delle cellule. Sono presenti nelle cellule di ogni organismo
Alberto Viale I CROMOSOMI DA MENDEL ALLA GENETICA AL DNA ALLE MUTAZIONI I cromosomi sono dei particolari bastoncelli colorati situati nel nucleo delle cellule. Sono presenti nelle cellule di ogni organismo
La rete dei centri di genetica e il futuro della medicina
 La rete dei centri di genetica e il futuro della medicina Rosario Casalone,, MD, PhD SSD Genetica Dipartimento Materno Infantile Azienda Ospedaliera-Polo Universitario Ospedale di Circolo e Fondazione
La rete dei centri di genetica e il futuro della medicina Rosario Casalone,, MD, PhD SSD Genetica Dipartimento Materno Infantile Azienda Ospedaliera-Polo Universitario Ospedale di Circolo e Fondazione
Gennaio 2014. Journal Club Febbraio 2014 Gruppo DI
 Gennaio 2014 2 Obiettivo dello studio: confrontare un ampio campione di soggetti con RASopatia con fratelli sani ed individui con Disturbo dello Spettro Autistico (ASD) al fine di 1. Determinare la prevalenza
Gennaio 2014 2 Obiettivo dello studio: confrontare un ampio campione di soggetti con RASopatia con fratelli sani ed individui con Disturbo dello Spettro Autistico (ASD) al fine di 1. Determinare la prevalenza
INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI
 SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI") INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene
INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene
PRODA Istituto di Diagnostica Clinica
 Test genetici per evidenziare il rischio di trombolfilia Il Fattore V della coagulazione è un cofattore essenziale per l attivazione della protrombina a trombina. La variante G1691A, definita variante
Test genetici per evidenziare il rischio di trombolfilia Il Fattore V della coagulazione è un cofattore essenziale per l attivazione della protrombina a trombina. La variante G1691A, definita variante
Docenti: Prof. Emilio Donti Sezione di Genetica Medica e Neuropsichiatria Infantile Dip. Scienze Chirurgiche e Biomediche Università degli Studi di
 GENETICA MEDICA Docenti: Prof. Emilio Donti Sezione di Genetica Medica e Neuropsichiatria Infantile Dip. Scienze Chirurgiche e Biomediche Università degli Studi di Perugia Centro Riferimento Regionale
GENETICA MEDICA Docenti: Prof. Emilio Donti Sezione di Genetica Medica e Neuropsichiatria Infantile Dip. Scienze Chirurgiche e Biomediche Università degli Studi di Perugia Centro Riferimento Regionale
Patologia del linguaggio in età evolutiva
 Patologia del linguaggio in età Ritardi e evolutiva disordini di acquisizione del linguaggio Disturbi del linguaggio Quadri clinici molto eterogenei : disturbi del linguaggio secondari le difficoltà linguistiche
Patologia del linguaggio in età Ritardi e evolutiva disordini di acquisizione del linguaggio Disturbi del linguaggio Quadri clinici molto eterogenei : disturbi del linguaggio secondari le difficoltà linguistiche
su registrazione frequenza cardiaca fetale registrazione contrazioni uterine Verifica dell effetto delle c.u. o dei MAF sulla FCF
 Nata x la diagnostica in travaglio di parto Sibasasu: su registrazione frequenza cardiaca fetale registrazione contrazioni uterine Verifica dell effetto delle c.u. o dei MAF sulla FCF Scaricato da www.sunhope.it
Nata x la diagnostica in travaglio di parto Sibasasu: su registrazione frequenza cardiaca fetale registrazione contrazioni uterine Verifica dell effetto delle c.u. o dei MAF sulla FCF Scaricato da www.sunhope.it
RIATTIVAZIONE EPIGENETICA DEL GENE FMR1
 RIATTIVAZIONE EPIGENETICA DEL GENE FMR1 Pietro Chiurazzi Istituto di Genetica Medica Università Cattolica del Sacro Cuore Sindrome X Fragile (OMIM #300624) La FXS è la causa più comune di ritardo psicomotorio
RIATTIVAZIONE EPIGENETICA DEL GENE FMR1 Pietro Chiurazzi Istituto di Genetica Medica Università Cattolica del Sacro Cuore Sindrome X Fragile (OMIM #300624) La FXS è la causa più comune di ritardo psicomotorio
La trasmissione delle malattie genetiche. Anna Onofri
 La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
Divisione riduzionale: meiosi
 Anomalie genetiche Divisione riduzionale: meiosi Nelle ovaie, con la meiosi si generano 4 nuclei da ogni ovogonio ma uno solo diventerà cellula-uovo mentre gli altri tre degenerano. Nei testicoli ogni
Anomalie genetiche Divisione riduzionale: meiosi Nelle ovaie, con la meiosi si generano 4 nuclei da ogni ovogonio ma uno solo diventerà cellula-uovo mentre gli altri tre degenerano. Nei testicoli ogni
S. I. E. D. P. GRUPPO DI STUDIO DELLA SINDROME DI TURNER
 S. I. E. D. P. GRUPPO DI STUDIO DELLA SINDROME DI TURNER PROTOCOLLO DI STUDIO SINDROME DI TURNER E PREVALENZA DI MATERIALE DERIVATIVO DEL CROMOSOMA Y E DI GONADOBLASTOMA CENTRO Data compilazione DATI ANAGRAFICI
S. I. E. D. P. GRUPPO DI STUDIO DELLA SINDROME DI TURNER PROTOCOLLO DI STUDIO SINDROME DI TURNER E PREVALENZA DI MATERIALE DERIVATIVO DEL CROMOSOMA Y E DI GONADOBLASTOMA CENTRO Data compilazione DATI ANAGRAFICI
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica
 Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Conoscenze, capacità, e comportamenti che ci si ripromette di trasmettere o sviluppare, con riferimento agli obiettivi di apprendimento
 Docente: HAYEK JOUSSEF Qualifica: DIRIGENTE MEDICO AOUS Insegnamento: NEUROPSICHIATRIA INFANTILE SSD: MED/39 Anno III o Descrivere l approccio clinico-strumentale al bambino con deficit intellettivo isolato
Docente: HAYEK JOUSSEF Qualifica: DIRIGENTE MEDICO AOUS Insegnamento: NEUROPSICHIATRIA INFANTILE SSD: MED/39 Anno III o Descrivere l approccio clinico-strumentale al bambino con deficit intellettivo isolato
EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI?
 EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI? CARATTERE AUTOSOMICO -codificato da geni su cromosomi non sessuali -non ci sono differenze di trasmissione
EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI? CARATTERE AUTOSOMICO -codificato da geni su cromosomi non sessuali -non ci sono differenze di trasmissione
DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE
 DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE Istituto di Genetica UNIVERSITA DEGLI STUDI DI UDINE Address: Istituto di Genetica DSTB - Università degli Studi di Udine P.le Kolbe,1-33100 Udine - ITALIA
DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE Istituto di Genetica UNIVERSITA DEGLI STUDI DI UDINE Address: Istituto di Genetica DSTB - Università degli Studi di Udine P.le Kolbe,1-33100 Udine - ITALIA
Il G-test aumenta il numero di malattie cromosomiche identificate con un semplice prelievo di sangue materno
 Il G-test aumenta il numero di malattie cromosomiche identificate con un semplice prelievo di sangue materno Roma, 28 gennaio 2016 Cresce in fretta il G-test, il test di screening non invasivo, effettuato
Il G-test aumenta il numero di malattie cromosomiche identificate con un semplice prelievo di sangue materno Roma, 28 gennaio 2016 Cresce in fretta il G-test, il test di screening non invasivo, effettuato
I marcatori molecolari. Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene
 I marcatori molecolari Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene Marcatori molecolari del DNA I marcatori molecolari sono sequenze di DNA
I marcatori molecolari Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene Marcatori molecolari del DNA I marcatori molecolari sono sequenze di DNA
Lo sviluppo del cancro è un processo complesso che coinvolge parecchi cambiamenti nella stessa cellula staminale. Poiché tutte le cellule staminali
 Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
SINDROMI POLIENDOCRINE AUTOIMMUNI (SPA)
") SINDROMI POLIENDOCRINE AUTOIMMUNI (SPA) Codice di esenzione: RCG030 Definizione. Con tale termine vengono definite varie sindromi cliniche caratterizzate da insufficienze funzionali a carico di una o più
SINDROMI POLIENDOCRINE AUTOIMMUNI (SPA) Codice di esenzione: RCG030 Definizione. Con tale termine vengono definite varie sindromi cliniche caratterizzate da insufficienze funzionali a carico di una o più
LA SINDROME DI WILLIAMS. a cura di Bruno Dallapiccola e Stefano Vicari. Genetica, clinica e riabilitazione
 Strumenti per il lavoro psico-sociale ed educativo LA SINDROME DI WILLIAMS Genetica, clinica e riabilitazione a cura di Bruno Dallapiccola e Stefano Vicari FrancoAngeli I lettori che desiderano informarsi
Strumenti per il lavoro psico-sociale ed educativo LA SINDROME DI WILLIAMS Genetica, clinica e riabilitazione a cura di Bruno Dallapiccola e Stefano Vicari FrancoAngeli I lettori che desiderano informarsi
PRODA Istituto di Diagnostica Clinica
 PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
Epidemiologia delle ipoacusie infantili
 Epidemiologia delle ipoacusie infantili 78 milioni di persone nel mondo presentano una ipoacusia moderata (>40 db) nell orecchio migliore e 364 milioni di persone hanno una ipoacusia lieve (26-40 db).
Epidemiologia delle ipoacusie infantili 78 milioni di persone nel mondo presentano una ipoacusia moderata (>40 db) nell orecchio migliore e 364 milioni di persone hanno una ipoacusia lieve (26-40 db).
MICRODELEZIONI DEL BRACCIO LUNGO DEL CROMOSOMA Y
 L infertilità è considerata dall Organizzazione Mondiale della Sanità una patologia. Per infertilità si intende l assenza di concepimento dopo 12/24 mesi di rapporti mirati non protetti. Il fenomeno dell
L infertilità è considerata dall Organizzazione Mondiale della Sanità una patologia. Per infertilità si intende l assenza di concepimento dopo 12/24 mesi di rapporti mirati non protetti. Il fenomeno dell
I BISOGNI EDUCATIVI SPECIALI
 I BISOGNI EDUCATIVI SPECIALI INCONTRO GENITORI CHIUSA PESIO Aprile 2015 Dott. ALESSANDRO MARANGI CORRELAZIONE (provvisoria) TRA MINORAZIONE e HANDICAP Legge 104/1992: è persona handicappata colui che presenta
I BISOGNI EDUCATIVI SPECIALI INCONTRO GENITORI CHIUSA PESIO Aprile 2015 Dott. ALESSANDRO MARANGI CORRELAZIONE (provvisoria) TRA MINORAZIONE e HANDICAP Legge 104/1992: è persona handicappata colui che presenta
INTOLLERANZA AL LATTOSIO: ESEMPIO DI BIODIVERSITA GENETICA
 INTOLLERANZA AL LATTOSIO: ESEMPIO DI BIODIVERSITA GENETICA I.P.S.I.A. Bettino Padovano Senigallia INDIRIZZO CHIMICO BIOLOGICO 2009/10 ALUNNO:MATTEO BARBARINI CLASSE: 5 TECNICO CHIMICO-BIOLOGICO DOCENTE:PROF.SSA
INTOLLERANZA AL LATTOSIO: ESEMPIO DI BIODIVERSITA GENETICA I.P.S.I.A. Bettino Padovano Senigallia INDIRIZZO CHIMICO BIOLOGICO 2009/10 ALUNNO:MATTEO BARBARINI CLASSE: 5 TECNICO CHIMICO-BIOLOGICO DOCENTE:PROF.SSA
Sintomi e segni. Disturbo. Sindrome. Disturbi Pervasivi dello Sviluppo
 Disturbi Pervasivi dello Sviluppo Annamaria Petito SSIS 400H Sintomi e segni Sono le informazioni che derivano da consapevoli sensazioni del paziente e sono le interpretazioni date dal medico agli elementi
Disturbi Pervasivi dello Sviluppo Annamaria Petito SSIS 400H Sintomi e segni Sono le informazioni che derivano da consapevoli sensazioni del paziente e sono le interpretazioni date dal medico agli elementi
Scala fenotipica. Dominante o recessivo? fenotipo. fenotipo. fenotipo A 1 A 1 A 2 A 2 A 1 A 2
 fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
RITARDO MENTALE. - Conoscere la definizione
 RITARDO MENTALE - Conoscere la definizione - Conoscere l epidemiologia e le cause - identificare I fattori prevenibili - Saper richiedere la consulenza genetica - Conoscere I rischi e i bisogni speciali
RITARDO MENTALE - Conoscere la definizione - Conoscere l epidemiologia e le cause - identificare I fattori prevenibili - Saper richiedere la consulenza genetica - Conoscere I rischi e i bisogni speciali
A cosa serve lo screening prenatale
 Screening e Diagnosi prenatale della sindrome di Down ed altre anomalie cromosomiche fetali Lo scopo di questa nota è fornire in maniera chiara, semplice e per quanto possibile esaustiva le informazioni
Screening e Diagnosi prenatale della sindrome di Down ed altre anomalie cromosomiche fetali Lo scopo di questa nota è fornire in maniera chiara, semplice e per quanto possibile esaustiva le informazioni
Prof. Pier Paolo Piccaluga Università di Bologna
 Prof. Pier Paolo Piccaluga Università di Bologna DNA: la molecola della vita L'acido desossiribonucleico (DNA) è un acido nucleico, presente nel nucleo delle cellule, che contiene le informazioni genetiche
Prof. Pier Paolo Piccaluga Università di Bologna DNA: la molecola della vita L'acido desossiribonucleico (DNA) è un acido nucleico, presente nel nucleo delle cellule, che contiene le informazioni genetiche
I Papillomavirus sono tutti uguali?
 Cos è il Papillomavirus? Il Papillomavirus è un microscopico nemico della tua salute. Attento, però, a non sottovalutare la pericolosità di questo microrganismo lungo solo 55 milionesimi di millimetro.
Cos è il Papillomavirus? Il Papillomavirus è un microscopico nemico della tua salute. Attento, però, a non sottovalutare la pericolosità di questo microrganismo lungo solo 55 milionesimi di millimetro.
Èun Associazione Amica di TELETHON.
 L Associazione ANGELI NOONAN Associazione Italiana Sindrome di Noonan Onlus è di recente costituzione: è nata il 15 marzo 2007. Si occupa dei bambini affetti dalla Sindrome di Noonan e dalle Sindromi Correlate
L Associazione ANGELI NOONAN Associazione Italiana Sindrome di Noonan Onlus è di recente costituzione: è nata il 15 marzo 2007. Si occupa dei bambini affetti dalla Sindrome di Noonan e dalle Sindromi Correlate
La Sindrome di Down Aspetti principali Dott.ssa Nicoletta Businaro
 Insegnamento Psicologia delle disabilità e dell integrazione (Prof.ssa Ottavia Albanese) La Sindrome di Down Aspetti principali Dott.ssa Nicoletta Businaro 12 Dicembre 2011 Per approfondimenti Dott.ssa
Insegnamento Psicologia delle disabilità e dell integrazione (Prof.ssa Ottavia Albanese) La Sindrome di Down Aspetti principali Dott.ssa Nicoletta Businaro 12 Dicembre 2011 Per approfondimenti Dott.ssa
www.fisiokinesiterapia.biz CARIOTIPO UMANO NORMALE E PATOLOGICO
 www.fisiokinesiterapia.biz CARIOTIPO UMANO NORMALE E PATOLOGICO CROMOSOMI Appaiono come corpi compatti solo nelle cellule in divisione, in particolare durante la metafase, quando possono essere identificati
www.fisiokinesiterapia.biz CARIOTIPO UMANO NORMALE E PATOLOGICO CROMOSOMI Appaiono come corpi compatti solo nelle cellule in divisione, in particolare durante la metafase, quando possono essere identificati
I Disturbi Specifici di Apprendimento. Brembati Federica Roberta Donini
 I Disturbi Specifici di Apprendimento Dott.ssa Brembati Federica Dott.ssa Roberta Donini Il disturbo specifico di apprendimento L uso del termine disturbo specifico dell apprendimento si riferisce a difficoltà
I Disturbi Specifici di Apprendimento Dott.ssa Brembati Federica Dott.ssa Roberta Donini Il disturbo specifico di apprendimento L uso del termine disturbo specifico dell apprendimento si riferisce a difficoltà
Le anomalie cromosomiche sono modificazioni del numero o della struttura dei cromosomi. Nella maggior parte dei casi si verificano durante la
 Le anomalie cromosomiche sono modificazioni del numero o della struttura dei cromosomi. Nella maggior parte dei casi si verificano durante la formazione dei gameti (ovociti e spermatozoi). Variazioni cromosomiche
Le anomalie cromosomiche sono modificazioni del numero o della struttura dei cromosomi. Nella maggior parte dei casi si verificano durante la formazione dei gameti (ovociti e spermatozoi). Variazioni cromosomiche
L'uso di SNP- array in diagnosi prenatale. Vanna Pecile IRCCS Burlo Garofolo Trieste
 L'uso di SNP- array in diagnosi prenatale Vanna Pecile IRCCS Burlo Garofolo Trieste Il cariotipo è in grado di evidenziare anomalie cromosomiche nel 8 35% di feti con malformazioni ecoevidenziate [Faas
L'uso di SNP- array in diagnosi prenatale Vanna Pecile IRCCS Burlo Garofolo Trieste Il cariotipo è in grado di evidenziare anomalie cromosomiche nel 8 35% di feti con malformazioni ecoevidenziate [Faas
La Citogenetica nella Diagnosi Prenatale
 La Citogenetica nella Diagnosi Prenatale Per diagnosi prenatale si intende l insieme delle indagini strumentali e di laboratorio finalizzate ad individuare determinate patologie su base : genetica infettiva
La Citogenetica nella Diagnosi Prenatale Per diagnosi prenatale si intende l insieme delle indagini strumentali e di laboratorio finalizzate ad individuare determinate patologie su base : genetica infettiva
BES Bisogni Educativi Speciali. Autismo. Lecco, 26 Febbraio 2015 Relatrice: Jessica Sala
 BES Bisogni Educativi Speciali Autismo Lecco, 26 Febbraio 2015 Relatrice: Jessica Sala DEFINIZIONE L autismo è una sindrome comportamentale causata da un disordine dello sviluppo, biologicamente determinato,
BES Bisogni Educativi Speciali Autismo Lecco, 26 Febbraio 2015 Relatrice: Jessica Sala DEFINIZIONE L autismo è una sindrome comportamentale causata da un disordine dello sviluppo, biologicamente determinato,
SEQUENZIAMENTO DEL DNA
 SEQUENZIAMENTO DEL DNA Il metodo di Sanger per determinare la sequenza del DNA Il metodo manuale La reazione enzimatica Elettroforesi in gel denaturante di poliacrilammide Autoradiografia Il metodo automatico
SEQUENZIAMENTO DEL DNA Il metodo di Sanger per determinare la sequenza del DNA Il metodo manuale La reazione enzimatica Elettroforesi in gel denaturante di poliacrilammide Autoradiografia Il metodo automatico
La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi
 La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi 1 Cromosomi omologhi I cromosomi in un corredo cromosomico diploide sono presenti come coppie di omologhi Negli animali le cellule
La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi 1 Cromosomi omologhi I cromosomi in un corredo cromosomico diploide sono presenti come coppie di omologhi Negli animali le cellule
Febbre Ricorrente Associata a NLRP12
 www.printo.it/pediatric-rheumatology/it/intro Febbre Ricorrente Associata a NLRP12 Versione 2016 1.CHE COS È LA FEBBRE RICORRENTE ASSOCIATA A NLRP12 1.1 Che cos è? La febbre ricorrente associata a NLRP12
www.printo.it/pediatric-rheumatology/it/intro Febbre Ricorrente Associata a NLRP12 Versione 2016 1.CHE COS È LA FEBBRE RICORRENTE ASSOCIATA A NLRP12 1.1 Che cos è? La febbre ricorrente associata a NLRP12
SINDROME CARDIOFACIALE DI CAYLER. Giangiorgio Crisponi
 SINDROME CARDIOFACIALE DI CAYLER ASIMMETRIA DELLA RIMA ORALE DURANTE IL PIANTO sindrome da delezione 22 q11 Giangiorgio Crisponi Cagliari Cagliari 2-3 Dicembre 2011 SINDROME CARDIOFACIALE DI CAYLER ASIMMETRIA
SINDROME CARDIOFACIALE DI CAYLER ASIMMETRIA DELLA RIMA ORALE DURANTE IL PIANTO sindrome da delezione 22 q11 Giangiorgio Crisponi Cagliari Cagliari 2-3 Dicembre 2011 SINDROME CARDIOFACIALE DI CAYLER ASIMMETRIA
DEFICIT ISOLATO ACTH
 DEFICIT ISOLATO ACTH Codice di esenzione: RC0010 Definizione. Il deficit isolato di ACTH è una rara causa di insufficienza surrenalica secondaria caratterizzata da una bassa o ridotta produzione di cortisolo
DEFICIT ISOLATO ACTH Codice di esenzione: RC0010 Definizione. Il deficit isolato di ACTH è una rara causa di insufficienza surrenalica secondaria caratterizzata da una bassa o ridotta produzione di cortisolo