Lezioni di genetica medica. Specializzazioni 2014
|
|
|
- Albano Carboni
- 8 anni fa
- Visualizzazioni
Transcript
1 Lezioni di genetica medica Specializzazioni 2014

2 Programma, parte 1 genetica umana generale Il genoma umano: geni ed organizzazione Next generation sequencing (NGS), l'exoma Eterogeneità clinica ed eterogeneità genetica Penetranza ed espressività, anticipazione Omozigosità ed eterozigosità composta Aploinsufficienza Meccanismo dello splicing e sue alterazioni Classi di mutazioni puntiformi, transizione e trasversione, conservative, missenso, nonsenso, nonstop Inserzioni, delezioni con frame shift e non, duplicazioni, conversione genica Significato patologico delle varie classi di variazioni del DNA: alleli equivalente, amorfo, ipomorfo, ipermorfo, neomorfo e antimorfo Nomenclatura delle variazioni genetiche e refertazione

3 Programma, parte 2: la consulenza e le cromosopatie La consulenza genetica: rischio riproduttivo dipendente ed indipendente dal partner Diagnostica prenatale e presintomatica L'analisi del cariotipo e i bandeggi, la FISH Cariotipo molecolare mediante arraycgh Aneuploidie negli aborti e rischio di ricorrenza Triploidia da doppio corredo paterno o materno, tetraploidia Il cromosoma X e la sua inattivazione, regioni PAR Trisomie autosomiche e dei cromosomi sessuali Le monosomie, la sindrome di Turner Delezioni cromosomiche, inversioni paracentriche e pericentriche Traslocazioni sbilanciate e bilanciate, robertsoniane, markers cromosomici Delezioni e duplicazioni submicroscopiche (s. di Williams, s. di DiGeorge, s. Cri du Chat)

4 Programma, parte 3 genetica medica mendeliana Malattie mendeliane monoalleliche con mutazioni de novo (craniosonostosi, acondroplasia) Malattie mendeliane monoalleliche a trasmissione autosomica dominante (neurofibromatosi, s. di Marfan, rene policistico, osteogenesi imperfetta) Malattie mendeliane monoalleliche legate al cromosoma X (distrofia muscolare di Duchenne e Becker, emofilia, ritardi mentali legati all X) Malattie mendeliane bialleliche a trasmissione autosomica recessiva (fibrosi cistica, alfa e beta talassemia, amiotrofia spinale, emocromatosi, glicogenosi)
 Malattie mendeliane bialleliche a trasmissione autosomica recessiva (fibrosi cistica, alfa e")
5 Programma, parte 4, eccezioni all eredità mendeliana Mutazioni dinamiche in regioni non codificanti (X fragile, distrofia miotonica) e codificanti (corea di Huntington, atassie spino cerebellari) Mutazioni in regioni cromosomiche con imprinting (Prader Willi, sindrome di Angelman, Beckwith Wiedemann, Silver Russel) disomia uniparentale Mutazioni del DNA mitocondriale (MERFF, MELAS, LHON, KS, s. di Leigh) Predisposizione genetica Caratteri multifattoriali studi GWAs

6 Testi consigliati Neri Genuardi Genetica umana e medica Editore Elsevier Masson Moncharmont Leonardi Patologia Generale Editore Idelson Gnocchi Strachan Read Genetica Molecolare Umana Editore Zanichelli Lewis Genetica Umana Editore Piccin Sito web (glossario)

7 portatori di un rischio riproduttivo indipendente dal partner portatori di una traslocazione cromosomica bilanciata (reciproca) scambio di materiale genetico tra cromosomi non omologhi frequenza 1/520 nati in genere è fenotipicamente normale Nell 1% dei casi vi èun fenotipo clinico traslocazione reciproca

8 traslocazioni reciproche (t) (rcp) nessuna macroregione cromosomica è apparentemente assente, ma solo trasferita su un altro cromosoma può essere interrotta la sequenza di un gene o di due geni (in eterozigosi) ècritico valutare i punti di rottura specie nelle traslocazioni de novo le delezioni possono essere apprezzate mediante array CGH si può produrre un gene di fusione tra due geni altrimenti separati, un evento che è comune nelle cellule maligne il problema maggiore èil rischio riproduttivo

9 traslocazioni bilanciate (meiosi e fertilizzazione) Segregazione alternata Traslocazione bilanciata Normale Segregazione adiacente 1 Traslocazione Traslocazione Segregazione adiacente 2 Trisomia Trisomia La formazione di tetravalenti aiuta a capire: con la segregazione alternata si formano gameti normali o con traslocazione bilanciata, mentre le segr. adiacenti 1 e 2 portano alla traslocazione sbilanciata o alla trisomia

10 traslocazioni robertsoniane (rob) rob coinvolgono i cromosomi acrocentrici 13, 14, 15, 21 e 22 nessuna regione cromosomica èassente, perché questi contengono un braccio corto privo di geni che può risultare perduto con la fusione dei bracci q di due cromosomi acrocentrici La più frequente traslocazione Robertsoniana èla rob(13q14q) che rappresenta il 75% di tutte le rob segue poi la rob(14q21q) e la rob(21q21q) si formano in genere durante la meiosi femminile e comportano infertilità maschile o abortività ripetuta.
 e la rob(21q21q) si formano in genere durante la meiosi femminile e comportano infertilità maschile o")
11 Percentuale alla nascita di figli con cariotipo sbilanciato da genitori con traslocazione robertsoniana t(13;14) M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5% t(21;21) M=F 100%
 F 15% M 2% t(21;22) F 10% M 5%")
12 Traslocazione sbilanciata maggiori sono le dimensioni cromosomiche, minore è la possibilità di una gravidanza a termine minori sono le dimensioni, maggiore èil rischio di un feto malformato Sesso del genitore donna>uomo (gli spermatozoi hanno il 7.5% di difetti contro l 1% degli oociti, ma sono selezionati) Il rischio aumenta se il difetto è stato accertato a partire da un figlio precedente con cariotipo sbilanciato
 Il rischio aumenta se il difetto è stato")
13 rischio di cariotipo sbilanciato Se non vi sono stati casi in famiglia e la madre è eterozigote per una traslocazione reciproca il rischio èil 7% Se non vi sono stati casi in famiglia e il padre è eterozigote per una traslocazione reciproca il rischio èil 3% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e la madre è eterozigote il rischio èil 14% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e il padre è eterozigote il rischio èl 8%

14 La tecnica del CGH (comparative genomic hybridization) permette l individuazione di sequenze delete o duplicate nel genoma da testare (red) mediante il confronto con un genoma di riferimento (green). Sono preparate due sonde fluorescenti di colore diverso che ibridano contemporaneamente sui cromosomi. Se in una regione cromosomica prevale il colore (green) relativo al genoma di controllo questo significa che il genoma da testare (red) ha una delezione in quella regione
 relativo al genoma di controllo questo significa che il genoma")
15

16 Formati dei Microarray Agilent 22K 44K 244K 1M Multiplex 11K 1.9K 105K 400K 44K 180K 15K 60K vecchi attuali nuovi

17 44,000 oligonucleotide with 8,769 interrogating probes (60 mer oligonucleotides) Agilent Technologies average spacing of probes across the DMD coding region of at least one every 144 base pairs digestion with AluI and Rsa I and labeling Agilent CGH Analytics V3.4s software F F

18 inversioni Le inversioni sono rare (meno di 1 caso su 1000) e a volte difficili da mettere in evidenza Possono essere semplici quando comprendono due punti di rottura su di un singolo cromosoma Sono pericentriche quando il segmento invertito contiene il centromero (es: 46, XX inv(3)p25q21) Le inversioni pericentriche dei cromosomi 1, 9, 16 e Y sono eteromorfismi citogenetici di normale riscontro in soggetti sani Le inversioni sono dette paracentriche se confinate ad uno dei due bracci (es: 46,XX. Inv(11)q21q23) L eterozigote per un inversione èun soggetto normale.

19 X-linked disorders DMD Duchenne Muscular Dystrophy - 1/3,500 boys Onset -- Early childhood - about 2 to 6 years Laboratory -- CK (50x to 1.000x), LDH5, ALT, AST, aldolase increase Symptoms -- Generalized weakness and muscle wasting affecting proximal limb muscles first. Calves often enlarged. Heart involvement Progression -- Disease progresses slowly but will affect all voluntary muscles. Survival possible beyond late twenties BMD Becker Muscular Dystrophy - 1/10,000 boys Onset -- Adolescence or adulthood Symptoms -- Almost identical to Duchenne but often much less severe. Heart involvement Progression -- Slower and more variable than DMD with survival well into mid to late adulthood

20 At least eight promoters are used to generate cell-specific expression of the dystrophin gene L = lymphocyte C = cortical M = muscle P = Purkinje R = retinal CNS = central nervous system S = Schwann G = general

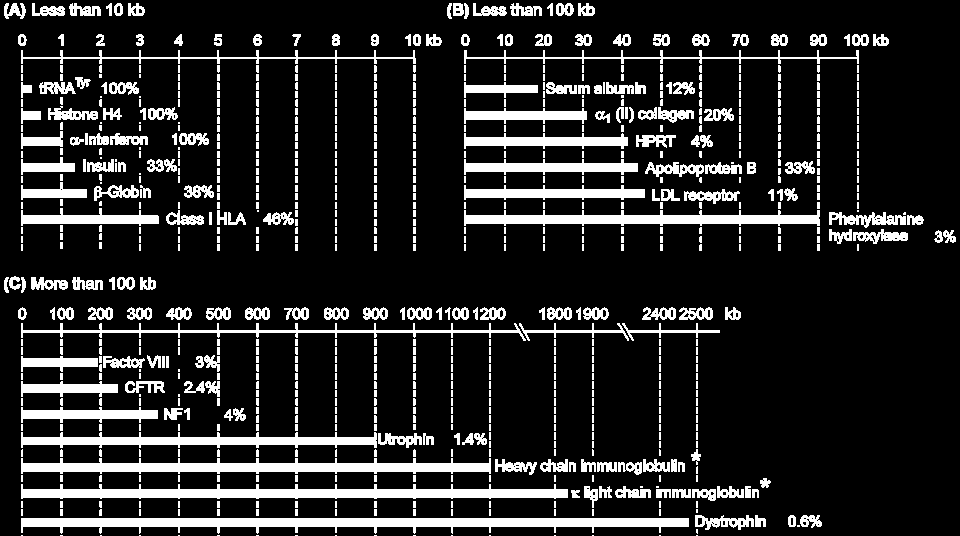
21

22
23
24
25
26 donna eterozigote per una traslocazione bilanciata X-autosoma
27
28 eterogeneità genetica Fenotipo clinico indistinguibile con pattern di trasmissione ereditaria differente: autosomico dominante, autosomico recessivo, X linked, o mitocondriale Esempio: le atassie cerebellari sono un gruppo di malattie neurodegenerative, in cui l'elemento dominante èla progressiva degenerazione cerebellare, con conseguente compromissione dell'equilibrio, dell andatura, della coordinazione dei movimenti degli arti e della parola
29 Proximal limb muscle involvement no apparent X-linked inheritance and generally milder severe LGMD (Duchennelike) or mild (Becker-like) great variability, ranging from severe forms with onset in the first decade and rapid progression to milder forms with later onset and a slower course
30 autosomal dominant autosomal dominant forms (LGMD1) are generally milder represent less than 10% of all LGMD marked heterogeneity for LGMD1, one gene per one single family
31
32 autosomal recessive autosomal recessive forms (LGMD2) have an average prevalence of 1:14,000-1:20,000 at birth frequency differences among countries this depends on higher carrier frequencies of single mutations, as 550delA for calpain 3 in Croatia, L276I for FKRP in Northern Europe, 521delT for gammasarcoglycan in Northern Africa At least 25% of families are excluded from any known locus and 40% of typical LGMD cases have no mutation in any known gene
33
34 aploinsufficienza insufficiente quantità di prodotto genico causata da una mutazione in eterozigosi la mutazione èdi tipo allele amorfo o ipomorfo colpisce geni per i quali il 50% di prodotto genico non è sufficiente a garantirne la funzione spesso un dosaggio preciso è richiesto ai fattori di trascrizione e alle molecole di segnale espressi nel corso dello sviluppo

35 50% A B La maggior parte dei geni si trova nella condizione A: il dosaggio genico critico è <50%. In tal caso, si osserva un fenotipo patologico solo se entrambi gli alleli sono colpiti, mentre in eterozigosi il fenotipo ènormale Pochi geni che si trovano nella condizione B: il dosaggio critico è >50%. Nelle delezioni genomiche submicroscopiche si osserva un fenotipo patologico in eterozigosi per aploinsufficienza
36 in caso di delezioni autosomiche in eterozigosi, molto spesso il dosaggio dimezzato non ècausa di malattia quando si osserva una sindrome da delezione, è risolutivo trovare la stessa sindrome causata da mutazioni puntiformi in uno solo dei geni se questa non si trova, la sindrome esiste solo come somma di più difetti
37
38 SMS del 17p11.2 sindrome di Smith-Magenis sporadica 1: , ma sottodiagnosticata ritardo mentale con ricerca dell attenzione e comportamenti autolesionistici (onicotillomania) disturbi del sonno faccia quadrata, brachicefalia, ipoplasia della parte centrale della faccia, brachidattilia voce grossa e profonda ipotonia, labbro superiore a tenda infezioni dell orecchio, anomalie oculari, anomalie cardiache e renali
39 ritardo nel linguaggio espressivo scarsa memoria a breve termine e difficoltà di sequenzialità sonno eccessivo diurno e interrotto notturno, per ciclo invertito della melatonina

 in")
40 SMS del 17p11.2 sindrome di Smith-Magenis 90% 10% ins/del puntiformi (frameshift) del gene RAI1 (retinoic acid induced 1) in eterozigosi nei casi senza delezione
41 Sindrome di DiGeorge
42 DiGeorge/velocardiofacciale La sindrome di DiGeorge del22q11.2 èla più frequente sindrome da microdelezione, con un incidenza di 1 su nati La delezione comprende 3Mb ed almeno 30 geni

43 le cellule della cresta neurale in migrazione contribuiscono alle strutture embrionali colpite nella s. di DiGeorge la figura rappresenta un embrione umano a 4 6 settimane di gestazione La migrazione delle cellule della cresta neurale dal cervello posteriore all arco branchiale / tasca faringea e del tratto di efflusso cardiaco è indicato dalle frecce. Le malformazioni associate a perturbazioni di questo processo sono elencati e queste coincidono sostanzialmente con quelle osservati nelle s. di DiGeorge 22q11 persistenza del dotto di Botallo (PDA), interruzione dell'arco aortico (IAA)
44 È caratterizzata da Anomalie cardiache T cell deficit palatoschisi anomalie facciali Ipocalcemia DiGeorge o velo/cardio/facciale Mutazioni puntiformi del gene TBX1 possono portare a questi 5 tratti fenotipici, ma non alle difficoltà nell apprendimento che èinvece frequente nella sindrome da delezione
45 Williams Beuren prevalenza alla nascita 1/7500 1/20.000, ma può non essere diagnosticata
46 Williams una delezione tipica
47 Williams genetica delezione de novo trasmissione autosomica dominante delezione di 1.6MB da 21 geni contigui in eterozigosi a 7q11.23 gene dell elastina LIM kinase 1 (LIMK1) CLIP-115 che lega i microtubuli Fattori di trascrizione GTF2I e GTF2IRD1 effetto posizionale su altri geni circostanti la delezione
48 Williams FISH delezione 7q11.23 rilevabile mediante FISH ma non cariotipo
49 Williams comportamento lieve o medio ritardo mentale (IQ tra 41 e 80) scarsa capacità di concentrazione ritardo nell apprendimento del linguaggio e poi esagerata loquacità personalità amichevole e affettuosa danno facilmente confidenza anche a sconosciuti ansietà, spesso preoccupati per il benessere altrui ipersensibilità ai suoni memoria visiva e uditiva spesso fuori dal comune ricordano persone, luoghi e motivi musicali predisposizione ad imparare le lingue e la musica
50 Williams aspetto e segni Faccia da elfo Occhi blu (77%) con pattern stellato dell iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa bocca larga e guance piene microdontia e micrognazia Statura 10 cm in meno del normale ipercalcemia stenosi periferica delle arterie polmonari stenosi aortica sopravalvolare
51 Williams foto
52 Williams foto
53 delezione de novo di circa 4MB Wolf Hirschhorn genetica le delezioni sono più frequenti nella linea germinale maschile trasmissione autosomica dominante Regione critica di 165 kb di molti geni contigui in eterozigosi a 4p16.3
54 Wolf Hirschhorn delezione a 4p16.3
55 Wolf-Hirschhorn Scarso accrescimento Ritardo mentale, ipotonia Labbro leporino Conformazione ad elmo di guerriero greco
56 Sindrome 5p- (cri du chat) 1: nati Pianto acuto e flebile Caratteristiche principali: Ritardo di crescita Microcefalia ed ipertelorismo Ipotonia, diastasi dei retti Deficit intellettivo e del linguaggio
57 MWS del 2q22 sindrome di Mowat Wilson sporadica 1: , ma sottodiagnosticata ritardo mentale gene ZEB2 lobo slargato e tipico
58 classificazione strutturale delle mutazioni 1. sostituzioni 2. piccole inserzioni, delezioni o inserzioni + delezioni contemporaneamente (indels) 3. riarrangiamenti genomici a due (delezioni, duplicazioni) o più punti di rottura (traslocazioni, inversioni ecc.) 4. copy number variations (CNV) a queste quattro classi appartengono in modo indistinguibile tanto le variazioni innocue quanto le mutazioni causative di malattia
59 numerazione dei nucleotidi Il nucleotide +1 èla A dell ATG codone di inizio della traduzione Il nucleotide che precede al 5' l ATG codone di inizio della traduzione èdenominato 1; non esiste una base 0 Il nucleotide che segue al 3' il codone di terminazione è denominato *1
60 sostituzioni le sostituzioni sono indicate dal carattere >. Ad esempio, 76A>C indica che in posizione 76 un adenina è sostituita da una citosina 88+1G>T (oppure IVS2+1G>T) indica che una guanina è sostituita da una timina in posizione +1 dell introne 2, posizionato tra i nucleotidi 88 e 89 del cdna 89 2A>C (oppure IVS2 2A>C) indica che un adenina è sostituita da una citosina in posizione 2 dell introne 2, posizionato tra i nucleotidi 88 e 89 del cdna
61
62 Mutationi osservate SNPs (polimorfismi) Teoricamente attese T>A or G, C>G or A G>T or C, A>T or G trasversioni T/A, C/G T/G, C/A trasversioni trasversioni transizioni T>C, C>T, G>A, A>G transizioni T/C, A/G transizioni 46,000 12,000,000 SEA 3057
63 Il meccanismo più comune di mutazione NH 2 NH 2 O N H 3 C N H 3 C NH N O metilazione N O deaminazione N O Cytosine 5-methylcytosine Thymine CG TG CG CA
64
65 mutazioni puntiformi missenso Le mutazioni missenso sono quelle in cui il cambiamento determina nel prodotto proteico la sostituzione di un aminoacido con un aminoacido differente Sebbene queste alterazioni generalmente non provochino conseguenze nella funzionalità della proteina (polimorfismi o varianti), ci sono casi in cui anche una minima alterazione può avere conseguenze gravi
66 acondroplasia nanismo dismorfico (1:35.000) arti corti e testa sproporzionatamente più grossa fronte prominente e naso appiattito altezza media 130 cm nei maschi 125 cm nelle femmine La mutazione èin eterozigosi Gly380Arg nel recettore 3 del "fibroblast growth factor" (FGFR3) a 4p16.3 autosomico dominante a penetranza completa
67
68 acondroplasia La mutazione conferisce una funzione aumentata al recettore dell'fgf (allele ipermorfo) che èuna tirosin chinasi di membrana In risposta all'fgf il recettore dimerizza e si fosforila trasducendo un segnale con la funzione di rallentare la proliferazione dei condrociti e quindi la crescita ossea Topi senza il gene FGF3R hanno ossa lunghe e vertebre allungate
69 ipocondroplasia L'ipocondroplasia ha caratteristiche simili all'acondroplasia, ma di gravità minore con un coinvolgimento craniofacciale inferiore. L'altezza può risultare ai limiti della norma e la malattia viene spesso non diagnosticata. L'ipocondroplasia èmeno omogenea: circa il 70% dei casi èdovuto alla sostituzione N540K del gene FGFR3, mentre non si conosce la mutazione nel restante 30%.
70 acrocefalosindattilia sindrome di Apert 1: alla nascita craniosinostosi, volta cranica a forma conica ipertensione endocranica ritardo mentale ipoplasia della parte centrale della faccia sindattilia delle dita delle mani e dei piedi sordità e atrofia ottica
71 acrocefalosindattilia sindrome di Apert tutti i pazienti hanno la stessa mutazione Apert (Cys755Gly) del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione èin eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Pfeiffer
72 sindrome di Pfeiffer alcuni pazienti hanno la mutazione Pfeiffer (Cys342Arg) del gene human fibroblast growth factor receptor 2 (FGFR2) altri la mutazione Pro252Arg in FGFR1 la mutazione èin eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Crouzon e Apert
73 disostosi cranio facciale sindrome di Crouzon alcuni pazienti hanno la mutazione (Cys342Tyr) del gene human fibroblast growth factor receptor 2 (FGFR2) la mutazione èin eterozigosi de novo cromosoma 10q26 la sindrome è allelica con Pfeiffer e Apert con alcune mutazioni in comune
74 mutazioni puntiformi nonsenso La mutazione nonsenso è quella in cui la modificazione nucleotidica provoca la creazione di un tripletta di stop, che blocca la sintesi della proteina prematuramente. In questo caso, la funzionalità della proteina dipenderà dalla posizione dello stop.
75 Tipi diversi di mutazioni nonsenso nei geni umani 15% 16% TAA TAG TGA 69% SEA 3063
76 mutazioni in eterozigosi di PAX3 Waardenburg sordità bilaterale (o deficit uditivo di vario livello) modifiche nella pigmentazione, sia dei capelli (albinismo parziale, in genere piebaldismo) sia della cute anomalie nello sviluppo dei tessuti derivati dalla cresta neurale lateralizzazione del canto mediale eterocromia, diverso colore degli occhi di solito uno marrone e l'altro blu
77 mutazioni eterozigoti di PAX3 Waardenburg
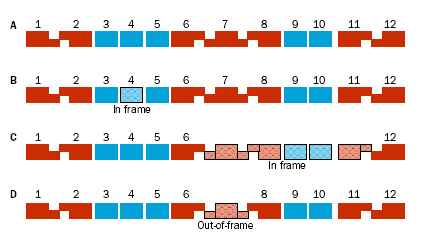
78 mutazioni frame-shift Le mutazioni frame shift o di slittamento del modulo di lettura consistono nell inserzione o delezione di un numero di nucleotidi non divisibile per 3 (1, 2, 4, 5, 7, 8, 10, ecc.) con conseguente sfasamento della cornice di lettura delle triplette dell'rna messaggero. Questa mutazione determina la traduzione non corretta della proteina a valle della mutazione.

79 Motivi classici di splicing
80 Nucleotidi intronici fiancheggianti inizio dell introne: il numero dell ultimo nucleotide dell esone che precede, il segno + e la posizione nell introne, ad esempio 77+1G, 77+2T, oppure quando il numero dell esone ènoto ed univoco IVS1+1G, IVS1+2T fine dell introne: il numero del primo nucleotide del esone seguente, il segno e la posizione a monte dell esone, ad esempio 78 2A, 78 1G, oppure quando il numero dell esone è noto ed univoco, IVS1 2A, IVS1 2G
81 Normal exon 5 ss IVS 3 ss exon 5 ss IVS 3 ss exon Splicing Abnormalities 5 ss mutation; exon skipping 3 ss mutation; exon skipping 5 ss mutation; use of cryptic 5 ss5 3 ss mutation; use of cryptic 3 ss3 Activation of cryptic 5 ss5 Activation of cryptic 5 ss 5 and use of cryptic 3 ss3 Splicing enhancer mutation Lariat structure branchpoint mutation
82 Progeria Hutchinson-Gilford invecchiamento precoce bassa statura, pelle rugosa calvizie, assenza di tessuto adiposo aterosclerosi ed infarto
83 Progeria Hutchinson-Gilford nuova mutazione in eterozigosi del gene lamina A la mutazione èin eterozigosi de novo cromosoma 1q23 La mutazione non cambia l'aminoacido glicina G608G, ma introduce un sito donor di splicing GGT che fa perdere 50 aminoacidi alla proteina sperimentazione con inibitori di farnesil trasferasi
84
85 Imprinting
86 Imprinting Nelle cellule germinali primordiali l imprinting viene cancellato del tutto e il DNA è demetilato Successivamente nella linea germinale maschile si determina un pattern di imprinting che in alcuni loci è complementare a quello della linea germinale femminile I cromosomi su cui avviene l imprinting (7, 11, 15) manterranno questo pattern e lo riprodurranno ad ogni mitosi Si potranno sempre distinguere l espressione genica del cromosoma materno e paterno
87 Disomia uniparentale Due copie dello stesso cromosoma sono ereditate dallo stesso genitore Spesso questo avviene attraverso un fenomeno transitorio di trisomia, seguito dalla perdita del cromosoma singolo e mantenimento del cromosoma doppio
88 70% dei casi delezione della regione cromosomica 15q11 q13, che è soggetta al fenomeno dell'imprinting del cromosoma paterno Il gene materno (l'unico espresso) può essere alterato con 4 meccanismi noti: delezione disomia uniparentale paterna difetti nell'imprinting mutazioni a carico del gene UBE3A (ubiquitin ligasi) La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi Angelman
89
90 "happy puppet syndrome" si può identificare in Cucciolo (Dopey) "addormentato", il più giovane dei nani che non ha mai imparato a parlare ritardo mentale con assenza del linguaggio, difficoltà nell'equilibrio, eccessivo buon umore Angelman
91 L'incidenza è 1/ nati crisi epilettiche e comunque alterazioni dell'eeg e microcefalia relativa Angelman
92 Prader-Willi iperfagia>obesità eccessiva assunzione di liquidi reazioni abnormi ai sedativi acromicria, criptorchidismo insensibilità al dolore, lesioni cutanee sbalzi di umore
93 Prader-Willi 1/15.000
94 Mutazioni dinamiche
95 Circa il 2% della popolazione ha un IQ<70 (ritardo mentale) il 15-20% di tutti I ritardi mentali sono attribuibili a geni del cromosoma X Il ritardo mentale legato al cromosoma X (XLMR) è geneticamente eterogeneo con 202 loci responsabili di forme che si sovrappongono clinicamente 46 geni sono stati a tutt oggi identificati il locus che contribuisce alla frazione maggiore causa la sindrome di Martin- Bell, oggi nota come sindrome dell X fragile
96 ritardo mentale: IQ tra 20 e 70 X fragile deficit di memoria a breve termine di informazioni complesse ritardo nel linguaggio ridotte abilità visuo-spaziali ipersensibilità agli stimoli iperattività con deficit di attenzione comportamento autistico Macrocefalia con fronte, mento e orecchie sporgenti Macroorchidismo (<30ml) dopo la pubertà Anomalie connettivali: prolasso della mitrale, lassità articolare, piede piatto Disfunzioni ipotalamiche?
97
98 Nel 1969 Lubs osservò una costrizione (marker X) sul braccio lungo del cromosoma X in quattro maschi affetti e tre carriers obbligate della stessa famiglia
99 Il sito fragile a Xq27.3 rottura o costrizione dei cromosomi in metafase che insorge quando le cellule sono esposte ad una perturbazione della replicazione del DNA siti fragili sono su tutti i cromosomi e prendono il nome della banda cromosomica, es fra(x)(q27.3) la nomenclatura HUGO chiama questo sito FRAXA, cioè il primo sito fragile identificato sul cromosoma X
100 Segregazione, paradosso di Sherman Il 20% dei maschi che portano l allele mutato sono normali (NTM) I Il 30% delle carrier presenta ritardo mentale perché è affetta? 1 perché non è affetto? II III 1 IV


101 Fragile X syndrome
102
103 X fragile al microscopio a forza atomica


104 Il gene FMR
105 Zhong et al. Am J Hum Genet controlli premutazioni
106 CpG island/5 UTR FMR1 gene Eag I EcoRI probe EcoRI 2.4kb (CGG) ~ 6 to 50 (CGG) 59 to ~ 200 (CGG) > 250 Methylation 2.8kb allele normale: stabile nella famiglia e nell individuo, instabile nella popolazione (polimorfismo) premutazione: instabile nella famiglia, stabile nell individuo mutazione piena: instabile nell individuo (mutazioni somatiche) reprime la trascrizione di FMR1
107 diagnosi di X Fragile : analisi mediante Southern blot di espansione e metilazione Rousseau et al. NEJM 1991
108 Premutazioni e mutazioni Le premutazioni si espandono quando sono trasmesse dalla madre La donna con premutazioni ha un maggiore rischio di menopausa precoce POF (premature ovarian failure) Il più corto allele descritto che in una sola generazione è diventato mutazione piena èdi 59 triplette Espansione stabile (CGG)9 AGG (CGG)9 AGG (CGG)9 Ha almeno 2 A che interrompono la serie di 9 triplette Espansione instabile (CGG)9 (CGG)9 (CGG)9 (CGG)9 NON ha A che interrompono la serie
109 Il gene FMR1 (fragile X mental retardation 1) è all interno di deserto genico: quindi il fenotipo NON è da geni contigui Mutazioni puntiformi o delezioni di FMR1 causano un fenotipo identico alle espansioni e questo dimostra che il ruolo del gene non è importante nelle prime fasi dello sviluppo, quando le triplette non sono ancora metilate
110 Trasmissione X fragile
111 cosa fa FMR1? FMR1 codifica per una RNA binding protein selettiva associata con i poliribosomi ed espressa nei neuroni nelle spine dendritiche regola la traduzione degli mrna, funzione cruciale per la plasticità sinaptica e la maturazione neuronale interagisce con gli mrna e con il pathway dei mirna Nell X fragile le spine dendritiche sono immature e lunghe
112 Spine dendritiche nel neocortex lunghe ed immature anche nel topo KO
113 Malattie da triplette ripetute Disease Gene Locus/Protein Repeat Location Fragile X syndrome Fragile XE syndrome Friedreich ataxia Myotonic dystrophy 1 Myotonic dystrophy 2 Spinobulbar muscular atrophy Huntington disease Dentatorubralpallidoluysian atrophy SCA type 1 SCA type 2 SCA type 3 (Machado-Joseph disease) SCA type 6 SCA type 7 SCA type 8 SCA type 12 Xq27.3/FMR-1 protein Xq28/FMR-2 protein 9q13-9q21.1/frataxin 19q13/myotonic dystrophy protein kinase 3q21 Xq13-Xq21/androgen receptor 4p16.3/huntington 12p13.31/atrophin-1 6p23/ataxin-1 12q24/ataxin-2 14q32.1/ataxin-3 19p13/α-1A (voltage-ependent calcium channel subunit) 3p12-3p13/ataxin-7 13q12/none identified 5q31-5q33 CGG GCC GAA CTG CCTG CTG Noncoding Noncoding Noncoding Noncoding Noncoding Coding Coding Coding Coding Coding Coding Coding Coding? Noncoding
114 Malattie da triplette ripetute non codificanti Sindrome del Xfragile Atassia di Friedreich Distrofia miotonica mutazione completa pre-mutazione normale CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG > GAA 7-22 esone GAA GAA GAA GAA GAA GAA GAA GAA GAA 200- > >2000 esone esone CTG CTG CTG CTG 5' UTR introne introne 3' UTR CTG CTG CTG CTG CTG CTG
115 anticipazione nella distrofia miotonica
116 distrofia miotonica DM1 fenomeno miotonico, difficoltà al rilasciamento muscolare dopo una contrazione ipotonia al volto, non debolezza importante cataratta precoce alterazioni ritmo cardiaco disfunzione tiroidea trasmissione autosomica dominante (1/8000) forma congenita con grave ipotonia neonatale
117 distrofia miotonica DM1 La distrofia miotonica di Steinert èla più comune distrofia muscolare dell adulto ècausata da un espansione CTG nel 3 UTR del gene DMPK (nell RNA CUG) a 19q13.3 presenta eredità autosomica dominante con anticipazione sono state identificate RNA binding proteins che interagiscono con l espansione CUG
118 distrofia miotonica DM2 cromosoma 3p21 un espansione simile nell introne 1 di un repeat CCUG nel gene ZNF9 (zinc finger protein 9) causa la distrofia miotonica 2 la DM2 èdetta anche distrofia miotonica prossimale
119 ammalato normale Malattie da triplette ripetute di poliglutammina ORF 5' UTR ' UTR normale ammalato Corea di Huntington Atassia spinocerebellare di tipo 2 Atassia spinocerebellare di tipo Atrofia dentatorubralepallidoluysiana Atrofia muscolare spinobulbare Malattia di Machado-Joseph Atassia spinocerebellare di tipo 6
120 Còrea di Huntington descritta da George Huntington nel 1872, è detta anche còrea che in greco indica la danza alla base vi è una degenerazione programmata geneticamente dei neuroni dei gangli basali (nuclei caudato e pallido) e della corteccia prevalenza di 1/10,000 e presenta il fenomeno dell'anticipazione si trasmette nel 97% dei casi come carattere autosomico dominante associato al gene huntingtina sul cromosoma 4p16.3 solo il 3% dei casi èdovuto a nuove mutazioni un'espansione dinamica della tripletta che codifica per la glutammina
121 quante glutammine? fino a 28 = numero max di per un soggetto non a rischio la malattia si potrebbe presentare alla generazione successiva (premutazione) oltre 39 il soggetto èconsiderato affetto anche se la patologia non si è ancora manifestata il test èin grado di prevedere che la patologia si manifesterà in futuro l'esecuzione in soggetti sani solleva problemi di natura etica
Genetica Medica corsi di laurea triennali
 Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
eterogeneità genetica
 eterogeneità genetica Fenotipo clinico indistinguibile con pattern di trasmissione ereditaria differente: autosomico dominante, autosomico recessivo, X-linked, o mitocondriale Esempio: le atassie cerebellari
eterogeneità genetica Fenotipo clinico indistinguibile con pattern di trasmissione ereditaria differente: autosomico dominante, autosomico recessivo, X-linked, o mitocondriale Esempio: le atassie cerebellari
Genetica Medica corsi di laurea triennali
 Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali
 Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
SCAMBI tra due coppie di cromosomi NON omologhi
 TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
5 modulo didattico - Patologia cromosomica.
 5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide
 Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
- Markers di anomalie cromosomiche. - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards)
 - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards)") - Markers di anomalie cromosomiche - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards) - Sindrome di Turner - Sindrome di Williams - Sindrome di Angelman
- Markers di anomalie cromosomiche - Trisomia 21 (sindrome di Down) - Trisomia 13 (sindrome di Patau) - Trisomia 18 (sindrome di Edwards) - Sindrome di Turner - Sindrome di Williams - Sindrome di Angelman
Anomalie cromosomiche
 Anomalie cromosomiche Alterazioni che interessano il DNA genomico determinando la perdita o l acquisizione di interi cromosomi o segmenti di essi. Se l alterazione è tale da poter essere visibile al microscopio
Anomalie cromosomiche Alterazioni che interessano il DNA genomico determinando la perdita o l acquisizione di interi cromosomi o segmenti di essi. Se l alterazione è tale da poter essere visibile al microscopio
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica
 Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Anomalie cromosomiche
 Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
Genetica Medica corsi di laurea triennali
 Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Genetica Medica corsi di laurea triennali Prof. Vincenzo Nigro Genetica Medica 1 anno, II semestre Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli programma del corso di genetica
Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
 Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked") Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Delezione. Duplicazione. Variazioni della struttura. Inversione. Traslocazione. Mutazioni cromosomiche. Nullisomia Monosomia Trisomia Tetrasomia
 Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
Lezioni di genetica medica. Specializzazioni 2014
 Lezioni di genetica medica Specializzazioni 2014 Programma, parte 1 genetica umana generale Il genoma umano: geni ed organizzazione Next generation sequencing (NGS), l'exoma Eterogeneità clinica ed eterogeneità
Lezioni di genetica medica Specializzazioni 2014 Programma, parte 1 genetica umana generale Il genoma umano: geni ed organizzazione Next generation sequencing (NGS), l'exoma Eterogeneità clinica ed eterogeneità
GENETICA MENDELIANA NELL UOMO
 GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
Gene Mutations and Disease
 Gene Mutations and Disease Mutazioni somatiche: Nuove mutazioni che insorgono casualmente nelle cellule somatiche o nella linea germinale di singoli individui. Le mutazioni germinali possono essere trasmesse
Gene Mutations and Disease Mutazioni somatiche: Nuove mutazioni che insorgono casualmente nelle cellule somatiche o nella linea germinale di singoli individui. Le mutazioni germinali possono essere trasmesse
LE MALATTIE GENETICHE CLASSE 3 C
 LE MALATTIE GENETICHE CLASSE 3 C Malattia causata da allele dominante Il nanismo acondroplastico è una malattia causata da un allele dominante; gli individui che ne sono affetti sono di statura molto bassa,
LE MALATTIE GENETICHE CLASSE 3 C Malattia causata da allele dominante Il nanismo acondroplastico è una malattia causata da un allele dominante; gli individui che ne sono affetti sono di statura molto bassa,
Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa
 Epigenetica ed espressione genica monoallelica Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa monoallelicamente,
Epigenetica ed espressione genica monoallelica Negli eucarioti, per la maggior parte dei geni, entrambe le copie sono espresse dalla cellula, ma una piccola classe di geni è espressa monoallelicamente,
VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI
 VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI Le alterazioni strutturali implicano cambiamenti di parti di cromosomi. Esistono 4 tipi di tali mutazioni: Delezione Duplicazione inversione Traslocazione Determinano
VARIAZIONI DELLA STRUTTURA DEI CROMOSOMI Le alterazioni strutturali implicano cambiamenti di parti di cromosomi. Esistono 4 tipi di tali mutazioni: Delezione Duplicazione inversione Traslocazione Determinano
Patologie da mutazioni dinamiche
 Patologie da mutazioni dinamiche Mutazioni dinamiche mutazioni progressive, nei tessuti e nelle generazioni, di ripetizioni instabili Alleli normali Pre-mutazioni Mutazioni Le alterazioni fenotipiche (patologia)
Patologie da mutazioni dinamiche Mutazioni dinamiche mutazioni progressive, nei tessuti e nelle generazioni, di ripetizioni instabili Alleli normali Pre-mutazioni Mutazioni Le alterazioni fenotipiche (patologia)
La mutazione è una modificazione della sequenza delle basi del DNA
 La mutazione è una modificazione della sequenza delle basi del DNA Le mutazioni sono eventi rari e importanti in quanto sono alla base dell evoluzione biologica Le mutazioni possono essere spontanee (dovute
La mutazione è una modificazione della sequenza delle basi del DNA Le mutazioni sono eventi rari e importanti in quanto sono alla base dell evoluzione biologica Le mutazioni possono essere spontanee (dovute
Eredità non mendeliana
 Eredità non mendeliana eredità extranucleare o citoplasmatica effetto materno eredità epigenetica (imprinting) anticipazione Eredità extranucleare eredità materna o paterna geni non nucleari: genomi mitocondriali
Eredità non mendeliana eredità extranucleare o citoplasmatica effetto materno eredità epigenetica (imprinting) anticipazione Eredità extranucleare eredità materna o paterna geni non nucleari: genomi mitocondriali
Mutazioni cromosomiche. Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo
 Mutazioni cromosomiche Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo Mutazioni cromosomiche Variazioni della struttura Variazioni
Mutazioni cromosomiche Le mutazioni cromosomiche sono la causa più frequente di aborto precoce e una importante causa di ritardo mentale nell uomo Mutazioni cromosomiche Variazioni della struttura Variazioni
4 modulo didattico - Modalità di trasmissione delle malattie
 4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
PRODA Istituto di Diagnostica Clinica
 PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI
 SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI") INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene eseguita su cellule
INFORMATIVA ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene eseguita su cellule
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali
 CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
= femmina. = maschio. = fenotipo banda bianca. = fenotipo pezzato. =fenotipo colore uniforme
 Test n.8 Dalle Olimpiadi delle Scienze Naturali 2002 PARTE TERZA Le 5 domande di questa parte riguardano il medesimo argomento e sono introdotte da un breve testo e da uno schema. In una razza bovina il
Test n.8 Dalle Olimpiadi delle Scienze Naturali 2002 PARTE TERZA Le 5 domande di questa parte riguardano il medesimo argomento e sono introdotte da un breve testo e da uno schema. In una razza bovina il
A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo
 2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
Scala fenotipica. Dominante o recessivo? fenotipo. fenotipo. fenotipo A 1 A 1 A 2 A 2 A 1 A 2
 fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136)
 (OMIM #607136)") ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136) Il gene implicato nella SCA17 è il gene TATA box-binding protein (TBP) che fa parte del complesso della RNA polimerasi II ed è essenziale per dare inizio
ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136) Il gene implicato nella SCA17 è il gene TATA box-binding protein (TBP) che fa parte del complesso della RNA polimerasi II ed è essenziale per dare inizio
Alberto Viale I CROMOSOMI
 Alberto Viale I CROMOSOMI DA MENDEL ALLA GENETICA AL DNA ALLE MUTAZIONI I cromosomi sono dei particolari bastoncelli colorati situati nel nucleo delle cellule. Sono presenti nelle cellule di ogni organismo
Alberto Viale I CROMOSOMI DA MENDEL ALLA GENETICA AL DNA ALLE MUTAZIONI I cromosomi sono dei particolari bastoncelli colorati situati nel nucleo delle cellule. Sono presenti nelle cellule di ogni organismo
La trasmissione delle malattie genetiche. Anna Onofri
 La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
La trasmissione dei caratteri ereditari. Le leggi di Mendel (1882-1884)
") La trasmissione dei caratteri ereditari Le leggi di Mendel (1882-1884) Le leggi di Mendel studiano la trasmissione di caratteri qualitativi prodotti da un singolo gene Procedimento sperimentale di Mendel
La trasmissione dei caratteri ereditari Le leggi di Mendel (1882-1884) Le leggi di Mendel studiano la trasmissione di caratteri qualitativi prodotti da un singolo gene Procedimento sperimentale di Mendel
Lezioni di genetica medica. Specializzazioni 2014
 Lezioni di genetica medica Specializzazioni 2014 Programma, parte 1 genetica umana generale Il genoma umano: geni ed organizzazione Next generation sequencing (NGS), l'exoma Eterogeneità clinica ed eterogeneità
Lezioni di genetica medica Specializzazioni 2014 Programma, parte 1 genetica umana generale Il genoma umano: geni ed organizzazione Next generation sequencing (NGS), l'exoma Eterogeneità clinica ed eterogeneità
EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI?
 EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI? CARATTERE AUTOSOMICO -codificato da geni su cromosomi non sessuali -non ci sono differenze di trasmissione
EREDITA MENDELIANA IL CARATTERE E TRASMESSO CON GLI AUTOSOMI O E ASSOCIATO AI CROMOSOMI SESSUALI? CARATTERE AUTOSOMICO -codificato da geni su cromosomi non sessuali -non ci sono differenze di trasmissione
GENETICA seconda parte
 GENETICA seconda parte I cromosomi sono lunghe molecole di una sostanza l acido desossiribonucleico. DNA Il DNA è una lunga catena fatta da due lunghi fili avvolti su se stessi a doppia elica. Sembra una
GENETICA seconda parte I cromosomi sono lunghe molecole di una sostanza l acido desossiribonucleico. DNA Il DNA è una lunga catena fatta da due lunghi fili avvolti su se stessi a doppia elica. Sembra una
GENETICA CITOGENETICA MEDICA
 CORSO INTEGRATO DI GENETICA E BIOLOGIA MOLECOLARE GENETICA A.A.2015/2016 Prof Alberto E.Turco Me 21.10.2015 Lezioni 15 e 16 CITOGENETICA MEDICA ANOMALIE CROMOSOMICHE (Chromosome Disorders) - Abortività
CORSO INTEGRATO DI GENETICA E BIOLOGIA MOLECOLARE GENETICA A.A.2015/2016 Prof Alberto E.Turco Me 21.10.2015 Lezioni 15 e 16 CITOGENETICA MEDICA ANOMALIE CROMOSOMICHE (Chromosome Disorders) - Abortività
Seconda Parte Specifica di scuola - Genetica medica - 31/07/2015
 Domande relative alla specializzazione in: Genetica medica Domanda #1 (codice domanda: n.311) : Qualora una donna risultasse positiva al test genetico per la ricerca di mutazioni del gene BRCA1, per quale
Domande relative alla specializzazione in: Genetica medica Domanda #1 (codice domanda: n.311) : Qualora una donna risultasse positiva al test genetico per la ricerca di mutazioni del gene BRCA1, per quale
Aggiornamenti in ambito genetico
 10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
Struttura e funzione dei geni. Paolo Edomi - Genetica
 Struttura e funzione dei geni 1 Il DNA è il materiale genetico La molecola di DNA conserva l informazione genetica: topi iniettati con solo DNA di batteri virulenti muoiono 2 Proprietà del DNA Il DNA presenta
Struttura e funzione dei geni 1 Il DNA è il materiale genetico La molecola di DNA conserva l informazione genetica: topi iniettati con solo DNA di batteri virulenti muoiono 2 Proprietà del DNA Il DNA presenta
Lo sviluppo del cancro è un processo complesso che coinvolge parecchi cambiamenti nella stessa cellula staminale. Poiché tutte le cellule staminali
 Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
Tecniche di bandeggio
 Tecniche di bandeggio sono sistemi di colorazione che conferiscono ai cromosomi caratteristici pattern di bande più o meno intense ogni cromosoma umano presenta un bandeggio (ossia una sequenza di bande)
Tecniche di bandeggio sono sistemi di colorazione che conferiscono ai cromosomi caratteristici pattern di bande più o meno intense ogni cromosoma umano presenta un bandeggio (ossia una sequenza di bande)
Componenti cellulari 24 2.3 Divisione e morte cellulare 38 Introduzione alla genetica 1 CERCASI DONATRICI DI OVULI
 L Autrice v Prefazione xiii L aspetto umano xvi Applicazioni della genetica umana xvii Il sistema Lewis di apprendimento guidato xviii P A R T E 1 Introduzione 1 A P I T O L O 1 2.2 omponenti cellulari
L Autrice v Prefazione xiii L aspetto umano xvi Applicazioni della genetica umana xvii Il sistema Lewis di apprendimento guidato xviii P A R T E 1 Introduzione 1 A P I T O L O 1 2.2 omponenti cellulari
1 a LEGGE DI MENDEL Legge della Segregazione
 IMPRINTING 1 a LEGGE DI MENDEL Legge della Segregazione I due membri di una coppia di alleli, durante la formazione dei gameti segregano in maniera indipendente, cioè in modo che metà dei gameti porti
IMPRINTING 1 a LEGGE DI MENDEL Legge della Segregazione I due membri di una coppia di alleli, durante la formazione dei gameti segregano in maniera indipendente, cioè in modo che metà dei gameti porti
-malattie monogeniche o mendeliane:
 Martedì 16 Febbraio è venuta nella nostra classe la dr.ssa Petrelli Maria a spiegarci le malattie sessualmente trasmissibili e ereditarie, sessualità e affettività. Ci ha spiegato la divisione delle cellule
Martedì 16 Febbraio è venuta nella nostra classe la dr.ssa Petrelli Maria a spiegarci le malattie sessualmente trasmissibili e ereditarie, sessualità e affettività. Ci ha spiegato la divisione delle cellule
MICRODELEZIONI DEL BRACCIO LUNGO DEL CROMOSOMA Y
 L infertilità è considerata dall Organizzazione Mondiale della Sanità una patologia. Per infertilità si intende l assenza di concepimento dopo 12/24 mesi di rapporti mirati non protetti. Il fenomeno dell
L infertilità è considerata dall Organizzazione Mondiale della Sanità una patologia. Per infertilità si intende l assenza di concepimento dopo 12/24 mesi di rapporti mirati non protetti. Il fenomeno dell
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI
 DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
Sindrome di Down. Aspetti genetici, fisici, motori e medici. www.fluitekruidje.com
 Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale. Dalla citogenetica convenzionale alla citogenetica molecolare!
 e diagnosi prenatale. Dalla citogenetica convenzionale alla citogenetica molecolare!") Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale Dalla citogenetica convenzionale alla citogenetica molecolare! CARIOTIPO STANDARD Identifica anomalie strutturali bilanciate e sbilanciate
Array-CGH(Comparative Genomic Hybridisation) e diagnosi prenatale Dalla citogenetica convenzionale alla citogenetica molecolare! CARIOTIPO STANDARD Identifica anomalie strutturali bilanciate e sbilanciate
PATOLOGIA CROMOSOMICA
 PATOLOGIA CROMOSOMICA CARIOTIPO UMANO NORMALE 4 6,X Y 4 6,X X CARIOTIPO UMANO NORMALE Cariotipo normale, giemsa CARIOTIPO UMANO NORMALE bande G C r o m o s o m a c o n b and e G t e lo m e r o c e ntr
PATOLOGIA CROMOSOMICA CARIOTIPO UMANO NORMALE 4 6,X Y 4 6,X X CARIOTIPO UMANO NORMALE Cariotipo normale, giemsa CARIOTIPO UMANO NORMALE bande G C r o m o s o m a c o n b and e G t e lo m e r o c e ntr
TARIFFARIO PRESTAZIONI LABORATORIO DI GENETICA MOLECOLARE, SEZIONE DI PSICOLOGIA CLINICA SANITARIA, DELLE ORGANIZZAZIONI E DEL BENESSERE
 TARIFFARIO PRESTAZIONI LABORATORIO DI GENETICA MOLECOLARE, SEZIONE DI PSICOLOGIA CLINICA SANITARIA, DELLE ORGANIZZAZIONI E DEL BENESSERE PRESTAZIONE IMPONIBILE IVA TOTALE Consulenza genetica 50,00 0,00
TARIFFARIO PRESTAZIONI LABORATORIO DI GENETICA MOLECOLARE, SEZIONE DI PSICOLOGIA CLINICA SANITARIA, DELLE ORGANIZZAZIONI E DEL BENESSERE PRESTAZIONE IMPONIBILE IVA TOTALE Consulenza genetica 50,00 0,00
LA GENETICA MENDELIANA NELLA SPECIE UMANA!
 LA GENETICA MENDELIANA NELLA SPECIE UMANA! ALBERI GENEALOGICI ACCOPPIAMENTO PROGENIE in ordine di nascita (da sx a dx) EREDITA AUTOSOMICA DOMINANTE AUTOSOMICA: il locus genico che controlla il carattere
LA GENETICA MENDELIANA NELLA SPECIE UMANA! ALBERI GENEALOGICI ACCOPPIAMENTO PROGENIE in ordine di nascita (da sx a dx) EREDITA AUTOSOMICA DOMINANTE AUTOSOMICA: il locus genico che controlla il carattere
OMOZIGOTE Dominante. OMOZIGOTE Recessivo ETEROZIGOTE
 GENI E CARATTERI EREDITARI I caratteri ereditari corrispondono a precisi tratti di DNA, i geni, che contengono le informazioni per la sintesi delle proteine. Ciascun gene occupa nel cromosoma una determinata
GENI E CARATTERI EREDITARI I caratteri ereditari corrispondono a precisi tratti di DNA, i geni, che contengono le informazioni per la sintesi delle proteine. Ciascun gene occupa nel cromosoma una determinata
GENOMA. c varia da pochi kb nei virus a milioni di kb in piante e animali
 GENOMA Insieme del materiale genetico presente in una cellula (DNA nucleare, plastidiale e mitocondriale) Contiene tutte le informazioni necessarie per consentire la vita alla cellula e all individuo Nei
GENOMA Insieme del materiale genetico presente in una cellula (DNA nucleare, plastidiale e mitocondriale) Contiene tutte le informazioni necessarie per consentire la vita alla cellula e all individuo Nei
CLASSIFICAZIONE STRUTTURALE DELLE MUTAZIONI
 CLASSIFICAZIONE STRUTTURALE DELLE MUTAZIONI 1. sostituzioni 2. piccole inserzioni, delezioni o inserzioni + delezioni contemporaneamente (indels) 3. riarrangiamenti genomici a due o più punti di rottura
CLASSIFICAZIONE STRUTTURALE DELLE MUTAZIONI 1. sostituzioni 2. piccole inserzioni, delezioni o inserzioni + delezioni contemporaneamente (indels) 3. riarrangiamenti genomici a due o più punti di rottura
La regolazione genica nei eucarioti
 La regolazione genica nei eucarioti Lic. Scientifico A. Meucci Aprilia Prof. Rolando Neri Differenziamento negli eucarioti pluricellulari Negli eucarioti le cellule specializzate dei vari tessuti contengono
La regolazione genica nei eucarioti Lic. Scientifico A. Meucci Aprilia Prof. Rolando Neri Differenziamento negli eucarioti pluricellulari Negli eucarioti le cellule specializzate dei vari tessuti contengono
Determinazione del sesso Cromosomi sessuali
 Determinazione del sesso Cromosomi sessuali Negli Eucarioti un cromosoma del sesso è un cromosoma presente in forme diverse nei due sessi. Uno è un cromosoma "X", l'altro strutturalmente e funzionalmente
Determinazione del sesso Cromosomi sessuali Negli Eucarioti un cromosoma del sesso è un cromosoma presente in forme diverse nei due sessi. Uno è un cromosoma "X", l'altro strutturalmente e funzionalmente
TEORIA CROMOSOMICA : ALLEGATI
 TEORIA CROMOSOMICA : ALLEGATI FIG. 2 a pag. 1 FIG. 5 a pag. 3 FIG. 7 a pag. 5 FIG. 9 a pag. 7 FIG. 3 e 4 a pag. 2 FIG. 6 a pag. 4 FIG. 8 a pag. 6 FIG. 10 e 11 a pag. 8 1 FIGURA 2 Perché sono tutti maschi
TEORIA CROMOSOMICA : ALLEGATI FIG. 2 a pag. 1 FIG. 5 a pag. 3 FIG. 7 a pag. 5 FIG. 9 a pag. 7 FIG. 3 e 4 a pag. 2 FIG. 6 a pag. 4 FIG. 8 a pag. 6 FIG. 10 e 11 a pag. 8 1 FIGURA 2 Perché sono tutti maschi
PRODA Istituto di Diagnostica Clinica
 Test genetici per evidenziare il rischio di trombolfilia Il Fattore V della coagulazione è un cofattore essenziale per l attivazione della protrombina a trombina. La variante G1691A, definita variante
Test genetici per evidenziare il rischio di trombolfilia Il Fattore V della coagulazione è un cofattore essenziale per l attivazione della protrombina a trombina. La variante G1691A, definita variante
Meiosi Genetica. Riproduzione sessuale Basi molecolari dell ereditarietà dei caratteri Variabilità genetica
 Meiosi Genetica Riproduzione sessuale Basi molecolari dell ereditarietà dei caratteri Variabilità genetica Batteri e altri organismi unicellulari si riproducono mediante divisione cellulare (riproduzione
Meiosi Genetica Riproduzione sessuale Basi molecolari dell ereditarietà dei caratteri Variabilità genetica Batteri e altri organismi unicellulari si riproducono mediante divisione cellulare (riproduzione
La genetica della Sindrome di Prader-Willi
 La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
Prima Legge di Mendel LEGGE DELLA SEGREGAZIONE IN PROPORZIONI UGUALI:
 Prima Legge di Mendel LEGGE DELLA SEGREGAZIONE IN PROPORZIONI UGUALI: Durante la meiosi, i membri di una coppia allelica si separano in modo simmetrico nelle uova e negli spermatozoi. Questa separazione
Prima Legge di Mendel LEGGE DELLA SEGREGAZIONE IN PROPORZIONI UGUALI: Durante la meiosi, i membri di una coppia allelica si separano in modo simmetrico nelle uova e negli spermatozoi. Questa separazione
RIATTIVAZIONE EPIGENETICA DEL GENE FMR1
 RIATTIVAZIONE EPIGENETICA DEL GENE FMR1 Pietro Chiurazzi Istituto di Genetica Medica Università Cattolica del Sacro Cuore Sindrome X Fragile (OMIM #300624) La FXS è la causa più comune di ritardo psicomotorio
RIATTIVAZIONE EPIGENETICA DEL GENE FMR1 Pietro Chiurazzi Istituto di Genetica Medica Università Cattolica del Sacro Cuore Sindrome X Fragile (OMIM #300624) La FXS è la causa più comune di ritardo psicomotorio
Dal DNA alle proteine: La trascrizione e la traduzione
 Dal DNA alle proteine: La trascrizione e la traduzione DNA RNA Trascrizione RNA PROTEINE Traduzione Dove avvengono? GLI EUCARIOTI I PROCARIOTI Cambell, Reece Biologia ZANICHELLI Trascrizione Sintesi di
Dal DNA alle proteine: La trascrizione e la traduzione DNA RNA Trascrizione RNA PROTEINE Traduzione Dove avvengono? GLI EUCARIOTI I PROCARIOTI Cambell, Reece Biologia ZANICHELLI Trascrizione Sintesi di
La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi
 La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi 1 Cromosomi omologhi I cromosomi in un corredo cromosomico diploide sono presenti come coppie di omologhi Negli animali le cellule
La divisione cellulare e la riproduzione degli organismi Parte II: Meiosi 1 Cromosomi omologhi I cromosomi in un corredo cromosomico diploide sono presenti come coppie di omologhi Negli animali le cellule
Perdita di funzione e fenotipo clinico
 Disordini genomici un disordine genomico submicroscopico è una patologia causata da Perdita Acquisizione Alterazione di uno o più geni contigui, le cui variazioni di dosaggio possono produrre effetti fenotipici.
Disordini genomici un disordine genomico submicroscopico è una patologia causata da Perdita Acquisizione Alterazione di uno o più geni contigui, le cui variazioni di dosaggio possono produrre effetti fenotipici.
Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche
 Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche La patogenesi di aborti dovuti ad anomalie cromosomiche è dovuta a: A) Ipoplasia placentare; B) Malformazioni
Definire i meccanismi con cui si verifica la correzione naturale delle anomalie cromosomiche La patogenesi di aborti dovuti ad anomalie cromosomiche è dovuta a: A) Ipoplasia placentare; B) Malformazioni
I marcatori molecolari. Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene
 I marcatori molecolari Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene Marcatori molecolari del DNA I marcatori molecolari sono sequenze di DNA
I marcatori molecolari Dipartimento di Scienze Agronomiche e Genetica Vegetale Agraria Corso di Genetica Agraria Giovanna Attene Marcatori molecolari del DNA I marcatori molecolari sono sequenze di DNA
GENETICA... lessico. Genetica: studio dei geni e dell'ereditarietà
 GENETICA... lessico Genetica: studio dei geni e dell'ereditarietà Geni: porzioni di DNA contenenti un'informazione che permette di decodificare una certa proteina. Es: gene che determina il colore dei
GENETICA... lessico Genetica: studio dei geni e dell'ereditarietà Geni: porzioni di DNA contenenti un'informazione che permette di decodificare una certa proteina. Es: gene che determina il colore dei
Come identificare i Cromosomi
 Come identificare i Cromosomi Fino al 1970, i cromosomi sono stati classificati in base alle dimensioni ed alla posizione del centromero. A I più grandi (metacentrici) B Grandi (submetacentrici) C Medi
Come identificare i Cromosomi Fino al 1970, i cromosomi sono stati classificati in base alle dimensioni ed alla posizione del centromero. A I più grandi (metacentrici) B Grandi (submetacentrici) C Medi
INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI
 SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI") INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene
INFORMATIVA E CONSENSO INFORMATO ALLA DIAGNOSI CITOGENETICA PRENATALE MOLECOLARE (ARRAY-CGH) SU CELLULE DEL LIQUIDO AMNIOTICO O VILLI CORIALI A. Finalità L indagine citogenetica fetale (o cariotipo) viene
Progetto sulle esostosi multiple
 Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
FINALITA : evidenziare la. cromosomiche fetali.
 Il Cariotipo Fetale Molecolare l (array-cgh) Il Cariotipo Fetale e TRADIZIONALE FINALITA : evidenziare la presenza di eventuali anomalie cromosomiche fetali. TECNICA: comporta la coltura delle cellule
Il Cariotipo Fetale Molecolare l (array-cgh) Il Cariotipo Fetale e TRADIZIONALE FINALITA : evidenziare la presenza di eventuali anomalie cromosomiche fetali. TECNICA: comporta la coltura delle cellule
Malattie genetiche. Il genoma umano e formato da 23 paia di cromosomi (1 paio di eterocromosomi, 22 paia di autosomi).
.") Malattie genetiche Il patrimonio genetico e contenuto nel DNA. Il genoma umano e formato da 23 paia di cromosomi (1 paio di eterocromosomi, 22 paia di autosomi). La sequenza di tutto il genoma umano e
Malattie genetiche Il patrimonio genetico e contenuto nel DNA. Il genoma umano e formato da 23 paia di cromosomi (1 paio di eterocromosomi, 22 paia di autosomi). La sequenza di tutto il genoma umano e
www.fisiokinesiterapia.biz LE LEGGI DI MENDEL
 www.fisiokinesiterapia.biz LE LEGGI DI MENDEL Gregor Johann Mendel (1822-1884) Comprese i principi che regolano la trasmissione dei caratteri ereditari alla progenie senza conoscere - l esistenza dei geni
www.fisiokinesiterapia.biz LE LEGGI DI MENDEL Gregor Johann Mendel (1822-1884) Comprese i principi che regolano la trasmissione dei caratteri ereditari alla progenie senza conoscere - l esistenza dei geni
ESTENSIONI DELLE LEGGI DI MENDEL
 ESTENSIONI DELLE LEGGI DI MENDEL Mendel rinuncia alle sue ricerche Mendel proseguì le sue ricerche su altre piante per ottenere conferme alle sue leggi, ma trovò tante e tali contraddizioni che (si dice)
ESTENSIONI DELLE LEGGI DI MENDEL Mendel rinuncia alle sue ricerche Mendel proseguì le sue ricerche su altre piante per ottenere conferme alle sue leggi, ma trovò tante e tali contraddizioni che (si dice)
LA SINDROME DI DOWN LA STORIA
 LA SINDROME DI DOWN LA STORIA La sindrome di Down, che è detta anche trisomia 21 o mongoloidismo, è una malattia causata dalla presenza di una terza copia del cromosoma 21; è la più comune anomalia cromosomica
LA SINDROME DI DOWN LA STORIA La sindrome di Down, che è detta anche trisomia 21 o mongoloidismo, è una malattia causata dalla presenza di una terza copia del cromosoma 21; è la più comune anomalia cromosomica
Unità Operativa di GENETICA MEDICA. Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara. Prestazioni richieste
 Unità Operativa di GENETICA MEDICA Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara Direttore: Prof.ssa Alessandra Ferlini www.ospfe.it/geneticamedica LABORATORIO DI GENETICA MOLECOLARE Responsabile:
Unità Operativa di GENETICA MEDICA Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara Direttore: Prof.ssa Alessandra Ferlini www.ospfe.it/geneticamedica LABORATORIO DI GENETICA MOLECOLARE Responsabile:
LA COSTRUZIONE DEL PEDIGREE NELLA GENETICA UMANA
 LA COSTRUZIONE DEL PEDIGREE NELLA GENETICA UMANA Un metodo di base di analisi genetica negli esseri umani è la costruzione di una storia familiare per seguire la trasmissione ereditaria di un carattere.
LA COSTRUZIONE DEL PEDIGREE NELLA GENETICA UMANA Un metodo di base di analisi genetica negli esseri umani è la costruzione di una storia familiare per seguire la trasmissione ereditaria di un carattere.
Fibrillina Sindrome di Marfan sindrome di Marfan sindrome di Marfan Sindrome di Marfan Fibrillina 1
 Genoma La determinazione e la conoscenza dell intera sequenza genomica è la condizione necessaria per comprendere la biologia di un determinato organismo Il genoma contiene le istruzioni (geni) per la
Genoma La determinazione e la conoscenza dell intera sequenza genomica è la condizione necessaria per comprendere la biologia di un determinato organismo Il genoma contiene le istruzioni (geni) per la
Il genoma dinamico: gli elementi trasponibili
 Il genoma dinamico: gli elementi trasponibili Anni trenta: studi sul mais ribaltano la visione classica secondo cui i geni si trovano solo in loci fissi sul cromosoma principale Esistono elementi genetici
Il genoma dinamico: gli elementi trasponibili Anni trenta: studi sul mais ribaltano la visione classica secondo cui i geni si trovano solo in loci fissi sul cromosoma principale Esistono elementi genetici
Emiatrofia cerebrale e microftalmia
 Emiatrofia cerebrale e microftalmia TROMBOSI TROMBOSI Prevenzione dell handicap congenito Nel 1 gruppo di casi genetici con rischio di ricorrenza, la prevenzione può attuarsi attraverso una corretta diagnosi
Emiatrofia cerebrale e microftalmia TROMBOSI TROMBOSI Prevenzione dell handicap congenito Nel 1 gruppo di casi genetici con rischio di ricorrenza, la prevenzione può attuarsi attraverso una corretta diagnosi
Mappatura genetica. alberi genealogici (pedigree) stima del rischio genetico (counseling) analisi di linkage (lod score) Paolo Edomi - Genetica
 stima del rischio genetico (counseling) analisi di linkage (lod score) Paolo Edomi - Genetica") Mappatura genetica alberi genealogici (pedigree) stima del rischio genetico (counseling) analisi di linkage (lod score) Alberi genealogici Simbologia negli alberi genealogici Eredità autosomica recessiva
Mappatura genetica alberi genealogici (pedigree) stima del rischio genetico (counseling) analisi di linkage (lod score) Alberi genealogici Simbologia negli alberi genealogici Eredità autosomica recessiva
LA GENETICA: DNA e RNA LA GENETICA. DNA e RNA. Prof. Daniele Verri
 LA GENETICA DNA e RNA Prof. Daniele Verri L'acido desossiribonucleico o deossiribonucleico (DNA) è un acido nucleico che contiene le informazioni necessarie per la formazione di RNA e proteine. LA GENETICA:
LA GENETICA DNA e RNA Prof. Daniele Verri L'acido desossiribonucleico o deossiribonucleico (DNA) è un acido nucleico che contiene le informazioni necessarie per la formazione di RNA e proteine. LA GENETICA:
GENETICA MENDELIANA. Per i suoi studi, Mendel utilizzò piante di pisello odoroso (Pisum sativum) Facilità di coltivazione. Disponibilità di varietà
 Facilità di coltivazione. Disponibilità di varietà") GENETICA: è la scienza che studia i caratteri ereditari degli organismi viventi, i meccanismi attraverso i quali si trasmettono ai discendenti e le modalità con cui si manifestano. La genetica moderna
GENETICA: è la scienza che studia i caratteri ereditari degli organismi viventi, i meccanismi attraverso i quali si trasmettono ai discendenti e le modalità con cui si manifestano. La genetica moderna
Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna
 Eredità mitocondriale Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna I mitocondri dello spermatozoo non entrano a far
Eredità mitocondriale Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna I mitocondri dello spermatozoo non entrano a far
La possibilita di conoscere i geni deriva dalla capacita di manipolarli:
 La possibilita di conoscere i geni deriva dalla capacita di manipolarli: -isolare un gene (enzimi di restrizione) -clonaggio (amplificazione) vettori -sequenziamento -funzione Il gene o la sequenza
La possibilita di conoscere i geni deriva dalla capacita di manipolarli: -isolare un gene (enzimi di restrizione) -clonaggio (amplificazione) vettori -sequenziamento -funzione Il gene o la sequenza
Dr.ssa Gabriella Silvestri Istituto di Neurologia, UCSC, Fondazione Policlinico A. Gemelli Roma Dr.ssa Marcella Masciullo IRCSS Santa Lucia, Roma
 Ereditarietà delle PSE e counselling genetico Dr.ssa Gabriella Silvestri Istituto di Neurologia, UCSC, Fondazione Policlinico A. Gemelli Roma Dr.ssa Marcella Masciullo IRCSS Santa Lucia, Roma Paraparesi
Ereditarietà delle PSE e counselling genetico Dr.ssa Gabriella Silvestri Istituto di Neurologia, UCSC, Fondazione Policlinico A. Gemelli Roma Dr.ssa Marcella Masciullo IRCSS Santa Lucia, Roma Paraparesi
Genetica. Mendel e la genetica
 Genetica Le leggi dell ereditarietà di Mendel Ereditarietà e cromosomi Estensioni della genetica mendeliana Applicazioni della genetica Genoma umano Mendel e la genetica Mendel 81822-1884), un monaco di
Genetica Le leggi dell ereditarietà di Mendel Ereditarietà e cromosomi Estensioni della genetica mendeliana Applicazioni della genetica Genoma umano Mendel e la genetica Mendel 81822-1884), un monaco di
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali. Silvia Sansavini
 CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel
 Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico. Volume sulle Disabilità intellettive, cap. 5.
 Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
Capitolo 3 Riproduzione e trasmissione dei cromosomi
 Capitolo 3 Riproduzione e trasmissione dei cromosomi 3.1 Nelle cellule somatiche del topolino domestico vi sono 40 cromosomi. a. Quanti cromosomi riceve dal padre? b. Quanti autosomi sono presenti nel
Capitolo 3 Riproduzione e trasmissione dei cromosomi 3.1 Nelle cellule somatiche del topolino domestico vi sono 40 cromosomi. a. Quanti cromosomi riceve dal padre? b. Quanti autosomi sono presenti nel
Analisi genetica nell uomo: alberi genealogici problemi interpretazione
 Analisi genetica nell uomo: alberi genealogici problemi interpretazione Tipi di ereditarietà monofattoriale autosomica dominante recessiva legata al sesso dominante recessiva Alberi genealogici o pedigrees
Analisi genetica nell uomo: alberi genealogici problemi interpretazione Tipi di ereditarietà monofattoriale autosomica dominante recessiva legata al sesso dominante recessiva Alberi genealogici o pedigrees
Le mutazioni Quando le informazioni sono errate
 Le mutazioni Quando le informazioni sono errate Lic. Scientifico A. Meucci Aprilia Prof. Rolando Neri Storia delle mutazioni Dal 1927 viene usato il concetto di mutazione così come è inteso oggi Il primo
Le mutazioni Quando le informazioni sono errate Lic. Scientifico A. Meucci Aprilia Prof. Rolando Neri Storia delle mutazioni Dal 1927 viene usato il concetto di mutazione così come è inteso oggi Il primo
Carpire il segreto della vita con l informatica Giosuè Lo Bosco Dipartimento di Matematica e Informatica, Università di Palermo, ITALY.
 Carpire il segreto della vita con l informatica Giosuè Lo Bosco Dipartimento di Matematica e Informatica, Università di Palermo, ITALY. Lezioni Lincee Palermo, 26 Febbraio 2015 Alla base della vita degli
Carpire il segreto della vita con l informatica Giosuè Lo Bosco Dipartimento di Matematica e Informatica, Università di Palermo, ITALY. Lezioni Lincee Palermo, 26 Febbraio 2015 Alla base della vita degli
I disturbi dell umore
 I DISTURBI DELL UMORE I disturbi dell umore Rappresentano la più comune patologia psichiatrica della età adulta Consistono in gravi sbalzi dell umore Comportano un rischio di suicidio del 19% Due categorie
I DISTURBI DELL UMORE I disturbi dell umore Rappresentano la più comune patologia psichiatrica della età adulta Consistono in gravi sbalzi dell umore Comportano un rischio di suicidio del 19% Due categorie
Patologia dell'asse ipotalamoipofisi-igf-cartilagine. Luca Taf Azienda USL 8 Arezzo UO Pediatria
 Patologia dell'asse ipotalamoipofisi-igf-cartilagine Luca Taf Azienda USL 8 Arezzo UO Pediatria Sistema complesso Azione del GH a livello cellulare Alcuni geni coinvolti nel processo di crescita GH-1 GHRH
Patologia dell'asse ipotalamoipofisi-igf-cartilagine Luca Taf Azienda USL 8 Arezzo UO Pediatria Sistema complesso Azione del GH a livello cellulare Alcuni geni coinvolti nel processo di crescita GH-1 GHRH