Overgrowth associated syndrome. Sindrome di Sotos. Epilessia e sindrome di Sotos
|
|
|
- Costanza Milano
- 5 anni fa
- Visualizzazioni
Transcript

1 Overgrowth associated syndrome Sindrome di Sotos Epilessia e sindrome di Sotos
2 Sindrome di Sotos È una rara malattia genetica, anche detta gigantismo cerebrale, caratterizzata prevalentemente da supercrescita (overgrowth) dismorfismi del volto tipici ritardo dello sviluppo psicomotorio Epidemiologia L esatta prevalenza è difficilmente valutabile a causa dell ampia variabilità nell espressione clinica è stimata approssimativamente 1/20000 nati.
3 EZIOPATOGENESI La sindrome è causata da mutazioni e delezioni del gene NSD1 (Nuclear receptor SET domain containing protein-1) localizzato sul braccio lungo del cromosoma 5 che codifica per una metiltrasferasi istonica implicata nella regolazione della trascrzione. Schematic representation of NSD1 Negli ultimi anni l analisi molecolare ha suggerito che anche altre mutazioni possano causare la sindrome in una percentuale piu bassa di casi (cromosomi X)
4 Eziopatogenesi Microdelezioni della regione cromosomica 5q35 ritardo mentale più severo e iperaccrescimento meno marcato Mutazioni puntiformi del gene NSD1 più frequente nelle popolazioni europee La maggiorparte delle mutazioni avvengono de novo, ma sono descritti rari casi familiari con ereditarietà autosomica dominante

5 Clinica Le manifestazioni cliniche coinvolgono diversi organi e apparati Accrescimento staturo-ponderale superiore alla norma soprattutto nei primi anni di vita circonferenza cranica DS peso +1 DS lunghezza +3.2 DS Alterazioni muscolo-scheletriche 84% età ossea 30% cifoscoliosi 20% iperlassità articolare piede piatto mani e piedi grandi

6 Clinica Alterazioni craniofacciali macrodolicocefalia fronte ampia e prominente impianto alto dei capelli (radi) costrizione bitemporale ipertelorismo downslanting delle rime palpebrali narici anteverse palato ogivale prognatismo

7 Clinica Alterazioni neuro-psicomotorie e comportamentali ipotonia alla nascita 30-50% convulsioni 45% EEG alterato 80ritardo motorio e del linguaggio QI variabile memoria a breve termine calcolo matematico tratti autistici Difetti oculari Glaucoma Nistagmo Distrofia/atrofia retinica Megalocornea, ipoplasia dell iride cataratta Strabismo e difetti rifrattivi nel 50% dei casi
 e giapponesi (35-50%) Apparato genitourinario reflusso")
8 Clinica Difetti cerebrali ventricoli laterali ipoplasia del corpo calloso leucomalacia periventricolare Difetti cardiaci DIA, DIV, PDA incidenza molto diversa tra popolazioni caucasice (8-20%) e giapponesi (35-50%) Apparato genitourinario reflusso vescicoureterale
 vie urinarie (20%) Endocrinopatie (14%) iper/ipotiroidismo intolleranza al")
9 Complicanze Neoplasie (2-3%) tumore di Wilms, epatocarcinoma, parotidi, osteocondroma, neuroblastoma, emangiomi, LNH, LLA, microcitoma polmonare, testicolari, fibromi ovarici Infezioni vie aeree superiori (72%) vie urinarie (20%) Endocrinopatie (14%) iper/ipotiroidismo intolleranza al glucosio

10 Diagnosi Non esistono criteri diagnostici ben definiti La clinica riveste un ruolo fondamentale e deve essere integrata da indagini radiologiche e riscontri anamnestici La detection rate dell analisi genetica è <100% la negatività dell analisi molecolare non consente di escludere definitivamente la diagnosi Possibile diagnosi prenatale se a conoscenza della mutazione NSD1 in uno dei genitori
11 Diagnosi Cardinal features present in 90% of cases Major features present in 15% of cases Characteristic facial appearance Advanced bone age Learning disability Cranial CT/MRI abnormalities Childhood overgrowth Poor feeding in infancy Neonatal jaundice Neonatal hypotonia Seizures Scoliosis Cardiac anomalies Renal anomalies Maternal pre-eclampsia Joint laxity/pes planus

12

13

14

15

16

17
18

19
20 Most SS patients have non progressive neurologic dysfunction and brain anomalies. Neurologic disturbances associated with SS are hypotonia, feeding difficulties, clumsiness, poor coordination, delayed language and motor development, behavioral anomalies, and seizures. The degree of learning impairment is estimated to be extremely variable, although it is present in almost all patients(tatton Brownet al.,2005).
21 Several studies affirm that seizures may appear in about 15 50% of patients, and they are estimated to be febrile in half of cases(tatton-brownet al.,2005;baujat&cormierdaire, 2007; Tatton-Brown & Rahman, 2007). A recent review of 21 SS adults described seizures as rare (9%) (Fickie et al., 2011). However, no data are available on whether patients with SS and febrile seizures (FS) later develop epilepsy,and no studies have investigated the electroclinical features and the long-term outcomes of epilepsy in these patients.
22 The aim of this study is to analyze the neurologic profile of 19 Caucasian patients with SS, with particular regard to epileptic phenotype and outcome.
23 Methods To be eligible for this study, patients had to have been clinically diagnosed with typical SS (e.g., presence of all the cardinal features in the same patient) (Tatton-Brown & Rahman, 2007) and had to present with at least one FS or afebrile seizure (AF).
24 Methods All the patients had to have undergone full neurologic examination, brain magnetic resonance imaging (MRI), EEG recording, and long-term clinical and EEG follow-up. Age at onset of seizures,seizure semiology,eeg characteristics,brain MRI findings,and therapy with antiepileptic drugs (AEDs) were analyzed for each case.
25 Genetic analysis of exons 2 23 of the NSD1 gene was performed in the reported patients by means of denaturing high performance liquid chromatography (HPLC; Transgenomics, Omaha,NE,U.S.A.),and direct sequencing of the chromatographic variant on 3130xl sequencer (Applied Biosystems, Foster City, CA, U.S.A.). Seizures were classified according to criteria of the International League Against Epilepsy (ILAE) (Engel, 2001). Written informed consent was obtained from parents or guardians of all recruited people.
26 Results Data from 19 Caucasian patients (15 male, four female) were collected from a cohort of patients referred to eight Italian Pediatric Neurology Divisions. The mean age at time of first evaluation was of 5 years 2 months (range 4 months 15 years), and the mean age at time of last follow-up was of11 years 2 months (range 6 20 years). Minimum follow-up period was of 5 years (range 5 19 years). Febrile and afebrile seizures were recorded in 11 and 15 patients, respectively; seven children with FS developed generalized and/or focal AF. Clinical, neurologic, neuroradiologic, and genetic features of the 19 patients are summarizedintable 1.
27
28 Febrile seizures, infantile spasms, absence, tonic clonic, and myoclonic seizures have been all associated with SS (Tatton-Brown & Rahman, 2007). Furthermore, a wide spectrum of behavioral and emotional disturbances may occur in SS patients, such as ADHD, aggressiveness, irritability, pyromania, social inhibition, psychosis, and autistic features (Mauceri et al., 2000; Tatton-Brown et al., 2005; de Boer et al., 2006). The patients with SS reported herein had FS and/or epilepsy.
29 Mean age of onset of FS was consistently before the onset of epilepsy (1 year 6 months vs. 5 years 5 months). The 64% of SS patients with FS developed epilepsy, a much higher Percentage if compared with patients with non syndromic FS (3 5%). We noticed that in some of the reported patients the diagnosis of SS was suspected only after they came to our attention for the first seizure, which was febrile in all cases.
30 Generalized tonic clonic seizures were the most frequent type among generalized AF, with a prevalence of 47%. Notably, the 40% of patients had temporal lobe seizures (TLS). Clinical symptomatology of TLS may include olfactory, gustatory, or auditory hallucinations, automatisms, fear, auras (e.g., abdominal aura) with/without behavioral arrest,deja vu or jamais vu like sensations.
31 In two patients, TLS presented with abdominal aura and bursts of aggressiveness, thus being initially confused with behavioral disorders. Ictal EEG recording allowed for the differential diagnosis between TLS and behavioral problems in these two patients.
32 Interictal EEG studies in patients with FS revealed normal background activity or anomalies fitting with the epileptic manifestations. Notably, anomalies in the temporal areas were recorded in a majority of patients with epilepsy at interictal EEG.
33 Seizures appeared easy to control with the common AEDs at standard doses, such as valproic acid, oxcarbamazepine, carbamazepine, lamotrigine, levetiracetam, topiramate.
34 In this case series, monotherapy with valproic acid was the most frequent used treatment. In general, AED administration resulted in progressive reduction and disappearance of seizures. Recurrence of seizures required dose correction; only two patients needed polytherapy
35 At the time of last follow-up, 4 of 15 epileptic patients discontinued AED therapy (mean seizure-free period: 5 years; seizure-free period range 2 10 years); the other 11 patients still receive AEDs with good clinical control in all cases but one(patient with periventricular nodularheterotopia).
36 The pattern of brain abnormalities in SS suggest delayed or disturbed maturation, in particular of midline structures. In a study of 40 SS cases, ventricular anomalies (90%), enlargement of supratentorial extracerebral fluid spaces (70%), and anomalies of corpus callosum (64%) and other midline structures (40%)were the typical findings.
37 Periventricular nodular heterotopias were reported in three patients (8%) (Schaeferet al.,1997).in thepresent series 17patients had central nervous system anomalies, which were similar to the previous alterations described in children with SS. Periventricular nodular heterotopia was observed in one case(5%). Ingeneral,we found alowe rincidence o fmidline brain anomalies compared with the series of Schaeferet al.
38 We cannot clarify the reason for the clinical and EEG involvement of temporal lobe in our patients, since no abnormalities were found in the temporal areas by MRI examination.
39 Furthermore, studies on the role of the NSD1 gene on the brain (or temporal lobe) development are missing, although it is known that the transcript of the NSD1 gene is expressed in brain tissue (Favarelli,2005).
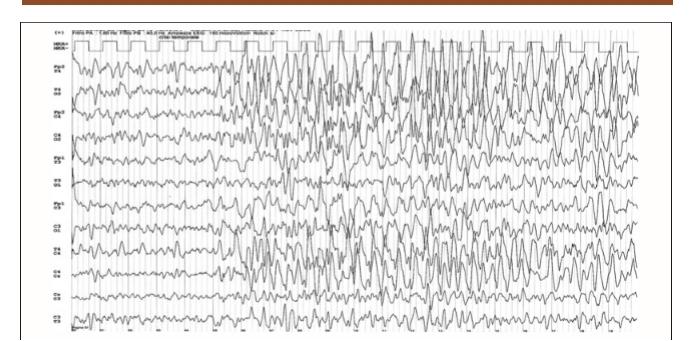
40 Ictal EEG
41 Conclusions This is the first study investigating the electroclinical features and the long-term follow-up of seizures in patients with SS. More than half of FS in these patients evolved to epilepsy.
42 Conclusions Generalized tonic clonic seizures were the more frequent type of generalized seizures, whereas TLS were recorded in the 40% of SS with epilepsy in this series.
43 Conclusions Because patients with SS may have both behavior disorders and seizures such as TLS, and since TLS may be confused with some behavior disorders, ictal EEG recording may be helpful for differential diagnosis. Seizure control seems to be easy to obtain with monotherapy, or, less frequently, polytherapy with AED. A careful neurologic evaluation is useful in SS because seizures are an important finding among people with this overgrowth syndrome.
44 NEWS NUOVI PATTERN RMN ED EEG
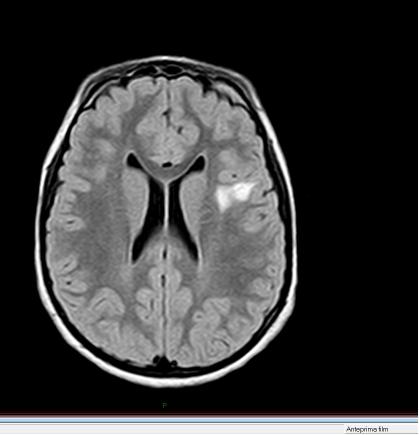

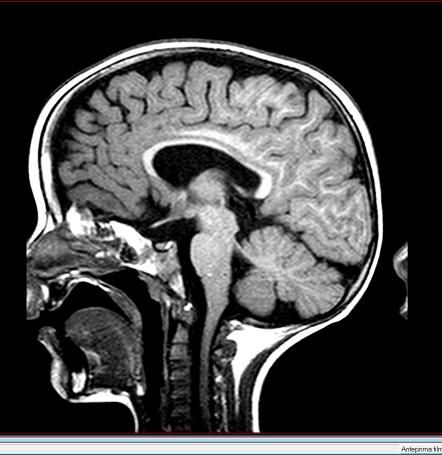
45 Rmn

46
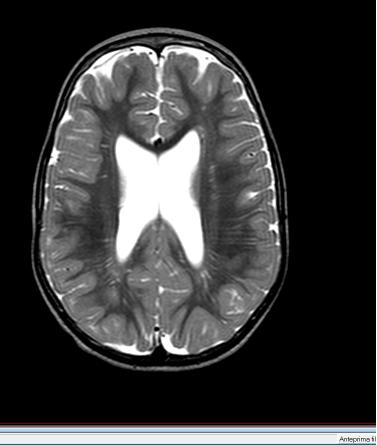
47
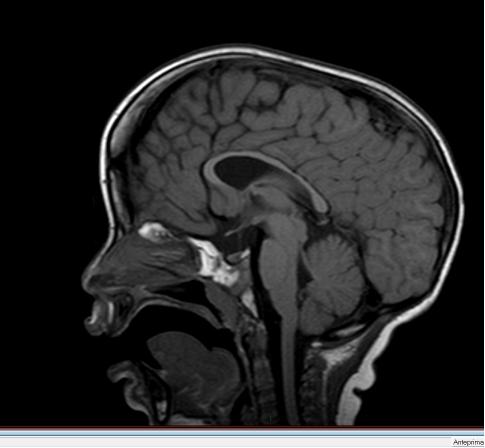
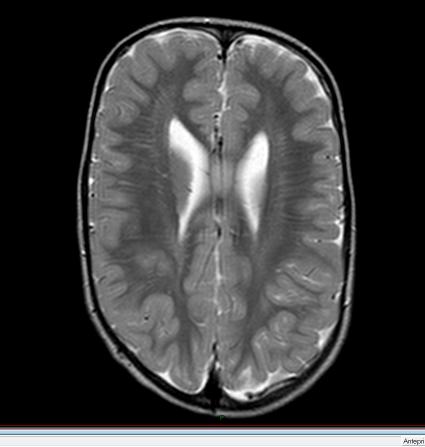
48
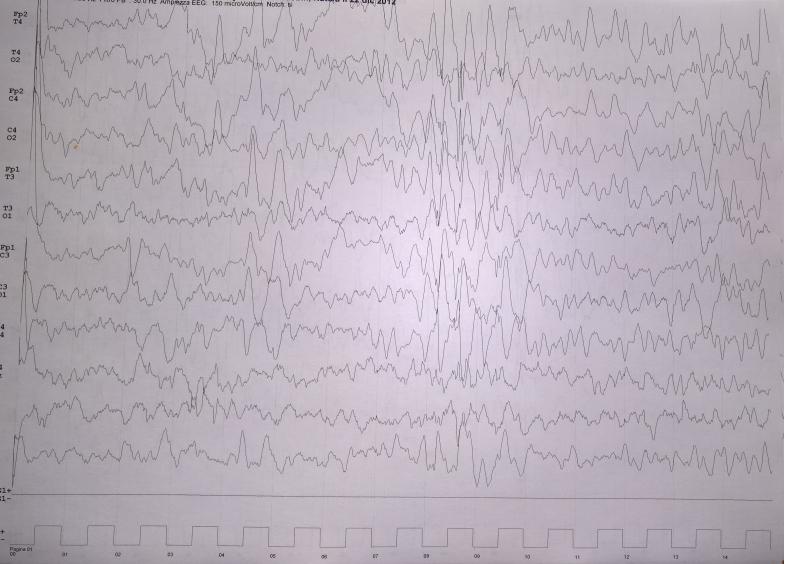
49
50 Diagnostic strategy when diagnosis of Sotos syndrome is suspected.
51 Diagnosi differenziale con tutte le condizioni associate a iperaccrescimento: Sindr. di Beckwith-Wiedemann Sindr. dell X-fragile Sindr. Weaver Sindr. Simpson-Golabi-Behmel Sindr. Gorlin Sindr. Bannayan-Riley-Ruvalcaba
52 Diagnosi differenziale Diagnostic flow chart for the differential diagnosis of tall stature and overgrowth syndromes.
53 Trattamento E sintomatico ed è rivolto alle patologie associate alla sindrome quali anomalie cardiache, epilessia, scoliosi, reflusso vescicoureterale, etc. Suggested approach to the evaluation and management of the individuals with Sotos syndrome.
54 PROSPETTIVE FUTURE CORRELAZIONE GENOTIPO FENOTIPO EEG E FUNZIONI COGNITIVE NUOVI PATTERN EEG FOLLOW-UP STUDI MULTICENTRICI
Trombosi in sedi atipiche: il ruolo delle nuove mutazioni. Walter Ageno Dipartimento di Medicina Clinica Università dell Insubria Varese
 Trombosi in sedi atipiche: il ruolo delle nuove mutazioni Walter Ageno Dipartimento di Medicina Clinica Università dell Insubria Varese Trombosi venose in sedi atipiche Trombosi venose in sedi diverse
Trombosi in sedi atipiche: il ruolo delle nuove mutazioni Walter Ageno Dipartimento di Medicina Clinica Università dell Insubria Varese Trombosi venose in sedi atipiche Trombosi venose in sedi diverse
LE CONVULSIONI FEBBRILI
 LE CONVULSIONI FEBBRILI FEDERICO VIGEVANO Divisione di Neurologia Ospedale Pediatrico Bambino Gesù - Le convulsioni febbrili rappresentano l evento convulsivo più frequente. Si calcola che circa il 4-5%
LE CONVULSIONI FEBBRILI FEDERICO VIGEVANO Divisione di Neurologia Ospedale Pediatrico Bambino Gesù - Le convulsioni febbrili rappresentano l evento convulsivo più frequente. Si calcola che circa il 4-5%
Le differenti manifestazioni cliniche della Sindrome di Tourette negli adulti
 Le differenti manifestazioni cliniche della Sindrome di Tourette negli adulti Massimo Pasquini diapartimento di neuroscience umane Sapienza Università di Roma Roma 20-04 - 2017 Sommario qnon abbiamo dati
Le differenti manifestazioni cliniche della Sindrome di Tourette negli adulti Massimo Pasquini diapartimento di neuroscience umane Sapienza Università di Roma Roma 20-04 - 2017 Sommario qnon abbiamo dati
Lesioni ghiandolari: il punto di vista molecolare. Giovanni Negri, Bolzano
 Lesioni ghiandolari: il punto di vista molecolare Giovanni Negri, Bolzano GiSCI, Firenze 2014 Lesioni ghiandolari: il punto di vista molecolare Quali sono i problemi maggiori nella diagnostica delle lesioni
Lesioni ghiandolari: il punto di vista molecolare Giovanni Negri, Bolzano GiSCI, Firenze 2014 Lesioni ghiandolari: il punto di vista molecolare Quali sono i problemi maggiori nella diagnostica delle lesioni
HODGKIN LYMPHOMA. IIa LINFOMA DI HODGKIN 0-14 ANNI. Schede specifiche per tumore. I tumori in Italia Rapporto AIRTUM 2012 TUMORI INFANTILI
 IIa LINFOMA DI HODGKIN HODGKIN LYMPHOMA -14 ANNI IIa LINFOMA DI HODGKIN -14 ANNI Il linfoma di Hodgkin rappresenta il 6% dei tumori registrati nel pool dei 31 registri della banca dati AIRTUM nel periodo
IIa LINFOMA DI HODGKIN HODGKIN LYMPHOMA -14 ANNI IIa LINFOMA DI HODGKIN -14 ANNI Il linfoma di Hodgkin rappresenta il 6% dei tumori registrati nel pool dei 31 registri della banca dati AIRTUM nel periodo
II LINFOMA E NEOPLASIE RETICOLOENDOTELIALI LYMPHOMA AND RETICULOENDOTHELIAL NEOPLASMS
 I tumori in Italia Rapporto AIRTUM 212 TUMORI INFANTILI II LINFOMA E NEOPLASIE RETICOLOENDOTELIALI LYMPHOMA AND RETICULOENDOTHELIAL NEOPLASMS -14 ANNI II LINFOMA -14 ANNI I linfomi rappresentano il 15%
I tumori in Italia Rapporto AIRTUM 212 TUMORI INFANTILI II LINFOMA E NEOPLASIE RETICOLOENDOTELIALI LYMPHOMA AND RETICULOENDOTHELIAL NEOPLASMS -14 ANNI II LINFOMA -14 ANNI I linfomi rappresentano il 15%
OSTEOSARCOMA. VIIIa OSTEOSARCOMA 0-14 ANNI. Schede specifiche per tumore. I tumori in Italia Rapporto AIRTUM 2012 TUMORI INFANTILI
 I tumori in Italia Rapporto AIRTUM TUMORI INFANTILI OSTEOSARCOMA - ANNI - ANNI INCIDENZA Tasso. Nella classe di età - anni, il tasso di incidenza standardizzato per età è di, casi per milione/anno (IC9%,-,)
I tumori in Italia Rapporto AIRTUM TUMORI INFANTILI OSTEOSARCOMA - ANNI - ANNI INCIDENZA Tasso. Nella classe di età - anni, il tasso di incidenza standardizzato per età è di, casi per milione/anno (IC9%,-,)
LE LINEE GUIDA: UTILIZZO RAZIONALE DELL ESPERIENZA ALTRUI
 LE LINEE GUIDA: UTILIZZO RAZIONALE DELL ESPERIENZA ALTRUI National Imaging Associates, 2005.keep in mind that the guidelines are not only intended to aid in the arrival at clinical consensus they may be
LE LINEE GUIDA: UTILIZZO RAZIONALE DELL ESPERIENZA ALTRUI National Imaging Associates, 2005.keep in mind that the guidelines are not only intended to aid in the arrival at clinical consensus they may be
RHABDOMYOSARCOMA. IXa RABDOMIOSARCOMA 0-14 ANNI. Schede specifiche per tumore. I tumori in Italia Rapporto AIRTUM 2012 TUMORI INFANTILI
 I tumori in Italia Rapporto AIRTUM TUMORI INFANTILI IXa RABDOMIOSARCOMA RHABDOMYOSARCOMA - ANNI IXa RABDOMIOSARCOMA - ANNI Il rabdomiosarcoma è il tipo istologico più frequente tra i tumori maligni dei
I tumori in Italia Rapporto AIRTUM TUMORI INFANTILI IXa RABDOMIOSARCOMA RHABDOMYOSARCOMA - ANNI IXa RABDOMIOSARCOMA - ANNI Il rabdomiosarcoma è il tipo istologico più frequente tra i tumori maligni dei
S. Bernasconi. Clinica Pediatrica. Università di Parma.
 S. Bernasconi Clinica Pediatrica Università di Parma sbernasconi@ao.pr.it Cianfarani 2008 2001 2003 Per poter accedere al trattamento con GH in individui nati SGA è necessario rispondere ai seguenti criteri:
S. Bernasconi Clinica Pediatrica Università di Parma sbernasconi@ao.pr.it Cianfarani 2008 2001 2003 Per poter accedere al trattamento con GH in individui nati SGA è necessario rispondere ai seguenti criteri:
Utilizzo e valutazione dell'offerta pubblica e privata di servizi radiotelevisivi nel 2017
 Utilizzo e valutazione dell'offerta pubblica e privata di servizi radiotelevisivi nel 2017 Rapporto finale Benjamin Fretwurst, Céline Murri, Thomas Friemel, Heinz Bonfadelli Panoramica grafica 3 Fretwurst/Murri/Friemel/Bonfadelli
Utilizzo e valutazione dell'offerta pubblica e privata di servizi radiotelevisivi nel 2017 Rapporto finale Benjamin Fretwurst, Céline Murri, Thomas Friemel, Heinz Bonfadelli Panoramica grafica 3 Fretwurst/Murri/Friemel/Bonfadelli
Epilessie farmacoresistenti Stefano Meletti Università di Modena e Reggio Emilia
 Epilessie farmacoresistenti Stefano Meletti Università di Modena e Reggio Emilia LA RICHIESTA DI COMPETENZA NEUROLOGICA NEL PROSSIOM FUTURO ROMA 13-14 APRILE 10 domande o concetti da sapere 1. Come si
Epilessie farmacoresistenti Stefano Meletti Università di Modena e Reggio Emilia LA RICHIESTA DI COMPETENZA NEUROLOGICA NEL PROSSIOM FUTURO ROMA 13-14 APRILE 10 domande o concetti da sapere 1. Come si
Epilessia. Federico Vigevano Dipartimento di Neuroscienze Ospedale Pediatrico Bambino Gesù Roma
 Epilessia Federico Vigevano Dipartimento di Neuroscienze Ospedale Pediatrico Bambino Gesù Roma 1 Epilessia Disordine del cervello caratterizzato da una persistente predisposizione a generare crisi epilettiche
Epilessia Federico Vigevano Dipartimento di Neuroscienze Ospedale Pediatrico Bambino Gesù Roma 1 Epilessia Disordine del cervello caratterizzato da una persistente predisposizione a generare crisi epilettiche
MODY (Maturity-Onset Diabetes of the Young)
") MODY (Maturity-Onset Diabetes of the Young) DM2 DM1 Difetti genetici della beta-cellula - Difetti genetici dell azione insulinica Sindromi genetiche rare associate al diabete Il MODY (Maturity-Onset Diabetes
MODY (Maturity-Onset Diabetes of the Young) DM2 DM1 Difetti genetici della beta-cellula - Difetti genetici dell azione insulinica Sindromi genetiche rare associate al diabete Il MODY (Maturity-Onset Diabetes
L endometriosi: il modello della paziente adolescente Stefano Ferrari
 L endometriosi: il modello della paziente adolescente Stefano Ferrari L ENDOMETRIOSI COME MALATTIA SOCIALE Atti Indagine Conoscitiva del Senato svolta dalla 12 Commissione Permanente del Senato (Igiene
L endometriosi: il modello della paziente adolescente Stefano Ferrari L ENDOMETRIOSI COME MALATTIA SOCIALE Atti Indagine Conoscitiva del Senato svolta dalla 12 Commissione Permanente del Senato (Igiene
Epilessia nelle anomalie del cromosoma 18
 Epilessia nelle anomalie del cromosoma 18 Robertino Dilena Servizio di Epilettologia e Neurofisiologia Pediatrica UOC Neurofisiopatologia Fondazione IRCCS Ca Granda Ospedale Maggiore Policlinico - Milano
Epilessia nelle anomalie del cromosoma 18 Robertino Dilena Servizio di Epilettologia e Neurofisiologia Pediatrica UOC Neurofisiopatologia Fondazione IRCCS Ca Granda Ospedale Maggiore Policlinico - Milano
LDL come target per il controllo della dislipidemia HIV-correlata
 LDL come target per il controllo della dislipidemia HIV-correlata EU vs USA: dove sta la verità? Dr Michele Bombelli Clinica Medica Università degli Studi Milano Bicocca Ospedale S.Gerardo, Monza LINEE
LDL come target per il controllo della dislipidemia HIV-correlata EU vs USA: dove sta la verità? Dr Michele Bombelli Clinica Medica Università degli Studi Milano Bicocca Ospedale S.Gerardo, Monza LINEE
Alla nascita riscontro di
 Maria Scavone Specializzanda in Pediatria Università degli Studi Magna Graecia di Catanzaro Antonio: Genitori non consanguinei, gravidanza normodecorsa Parto spontaneo eutocico, 38 W + 1 g APGAR 1 10 5
Maria Scavone Specializzanda in Pediatria Università degli Studi Magna Graecia di Catanzaro Antonio: Genitori non consanguinei, gravidanza normodecorsa Parto spontaneo eutocico, 38 W + 1 g APGAR 1 10 5
Single-rate three-color marker (srtcm)
") 3. Markers Pag. 1 The Single Rate Three Color Marker (srtcm) can be used as component in a Diffserv traffic conditioner The srtcm meters a traffic stream and marks its packets according to three traffic
3. Markers Pag. 1 The Single Rate Three Color Marker (srtcm) can be used as component in a Diffserv traffic conditioner The srtcm meters a traffic stream and marks its packets according to three traffic
TITLE. Hip Chondral Defects: How to Treat Them. AUTHOR. Dr. Andrea Fontana. Istituto Clinico Città Studi Milano
 TITLE Hip Chondral Defects: How to Treat Them. AUTHOR Dr. Andrea Fontana. Istituto Clinico Città Studi Milano TEXT (SUMMARY) ENGLISH VERSION Since 1995 on a total of 778 hip arthroscopies, the incidence
TITLE Hip Chondral Defects: How to Treat Them. AUTHOR Dr. Andrea Fontana. Istituto Clinico Città Studi Milano TEXT (SUMMARY) ENGLISH VERSION Since 1995 on a total of 778 hip arthroscopies, the incidence
Diagnosi precoce: utopia o realtà?
 Diagnosi precoce: utopia o realtà? Valter Torri Laboratorio di Metodologia per la Ricerca Clinica Dipartimento di Oncologia Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano è il 3% del totale
Diagnosi precoce: utopia o realtà? Valter Torri Laboratorio di Metodologia per la Ricerca Clinica Dipartimento di Oncologia Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano è il 3% del totale
19-20 Novembre 2014, Pisa Scuola Medica della Facoltà di Medicina. Co.Me.Lab 2014
 19-20 Novembre 2014, Pisa Scuola Medica della Facoltà di Medicina Co.Me.Lab 2014 Aldo Clerico Scuola Superiore Sant Anna Fondazione Regione Toscana G. Monasterio clerico@ftgm.it INTRODUZIONE Il BNP è un
19-20 Novembre 2014, Pisa Scuola Medica della Facoltà di Medicina Co.Me.Lab 2014 Aldo Clerico Scuola Superiore Sant Anna Fondazione Regione Toscana G. Monasterio clerico@ftgm.it INTRODUZIONE Il BNP è un
Gli Antidepressivi nella depressione maggiore. Trieste, 26 Febbraio 2009
 Gli Antidepressivi nella depressione maggiore Trieste, 26 Febbraio 2009 PRINCIPI GENERALI (I) Gli antidepressivi dovrebbero essere prescritti nei soggetti con depressione maggiore di intensità media-grave
Gli Antidepressivi nella depressione maggiore Trieste, 26 Febbraio 2009 PRINCIPI GENERALI (I) Gli antidepressivi dovrebbero essere prescritti nei soggetti con depressione maggiore di intensità media-grave
R.M.Dorizzi, Strumenti per interpretare gli esami di laboratorio? Forlì, 9 ottobre 2012
 1 Half of what you are taught as medical students will in 10 years have been shown to be wrong. And the trouble is, none of your teachers know which half. Sydney Burwell, Dean Harvard Medical School 1935-1949
1 Half of what you are taught as medical students will in 10 years have been shown to be wrong. And the trouble is, none of your teachers know which half. Sydney Burwell, Dean Harvard Medical School 1935-1949
Graphs: Cycles. Tecniche di Programmazione A.A. 2012/2013
 Graphs: Cycles Tecniche di Programmazione Summary Definitions Algorithms 2 Definitions Graphs: Cycles Cycle A cycle of a graph, sometimes also called a circuit, is a subset of the edge set of that forms
Graphs: Cycles Tecniche di Programmazione Summary Definitions Algorithms 2 Definitions Graphs: Cycles Cycle A cycle of a graph, sometimes also called a circuit, is a subset of the edge set of that forms
Filippo Muratori. diagnosi
 Filippo Muratori Il diritto alla diagnosi N = 10.138 children(7-9 years old attending school) 9.830 in public school+ 308 in private school). 239 certified children 61 ASD certified children 1/161 (0.62%)
Filippo Muratori Il diritto alla diagnosi N = 10.138 children(7-9 years old attending school) 9.830 in public school+ 308 in private school). 239 certified children 61 ASD certified children 1/161 (0.62%)
I principali bias. bias:
 I principali bias bias: any systematic error in an epidemiologic study that results in an incorrect estimate of the association between exposure and risk of disease What is Bias Any trend in the collection,
I principali bias bias: any systematic error in an epidemiologic study that results in an incorrect estimate of the association between exposure and risk of disease What is Bias Any trend in the collection,
LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI
 Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
TRATTAMENTO CHIRURGICO dell EPILESSIA: ANALISI di 1223 PAZIENTI con ESORDIO di MALATTIA in ETÀ PEDIATRICA
 UNIVERSITÀ DEGLI STUDI DIPAVIA FACOLTÀ DI MEDICINA E CHIRURGIA Neuropsichiatria Infantile Direttore: Chiar.mo Prof. U. Balottin TRATTAMENTO CHIRURGICO dell EPILESSIA: ANALISI di 1223 PAZIENTI con ESORDIO
UNIVERSITÀ DEGLI STUDI DIPAVIA FACOLTÀ DI MEDICINA E CHIRURGIA Neuropsichiatria Infantile Direttore: Chiar.mo Prof. U. Balottin TRATTAMENTO CHIRURGICO dell EPILESSIA: ANALISI di 1223 PAZIENTI con ESORDIO
Filippo Caraci & Filippo Drago
 Corso di Laurea Magistrale in Psicologia Laboratorio di Psicofarmacologia Università degli Studi di Catania Uso off-label degli antipsicotici di seconda generazione Filippo Caraci & Filippo Drago Uso off-label
Corso di Laurea Magistrale in Psicologia Laboratorio di Psicofarmacologia Università degli Studi di Catania Uso off-label degli antipsicotici di seconda generazione Filippo Caraci & Filippo Drago Uso off-label
Combinazioni serie HL-MHL + MHL. Sono disponibili varie combinazioni tra e riduttori coassiali serie MHL (2 stadio).
.") Combinazioni tra riduttori serie HL-MHL e MHL Possible combined units of helical inline gearboxes HL-MHL+MHL Combinazioni serie HL-MHL + MHL Sono disponibili varie combinazioni tra riduttori coassiali
Combinazioni tra riduttori serie HL-MHL e MHL Possible combined units of helical inline gearboxes HL-MHL+MHL Combinazioni serie HL-MHL + MHL Sono disponibili varie combinazioni tra riduttori coassiali
Salvatore De Fazio MD, PhD SerT Mesagne Presidente SITD Puglia
 Salvatore De Fazio MD, PhD SerT Mesagne Presidente SITD Puglia REINSTATEMENT DATI PRECLINICI: diminuzione del reinstatement da cocaina, nicotina, eroina per la somministrazione cronica (Reichel
Salvatore De Fazio MD, PhD SerT Mesagne Presidente SITD Puglia REINSTATEMENT DATI PRECLINICI: diminuzione del reinstatement da cocaina, nicotina, eroina per la somministrazione cronica (Reichel
Scheduling. Scheduler. Class 1 Class 2 Class 3 Class 4. Scheduler. Class 1 Class 2 Class 3 Class 4. Scheduler. Class 1 Class 2 Class 3 Class 4
 Course of Multimedia Internet (Sub-course Reti Internet Multimediali ), AA 2010-2011 Prof. 4. Scheduling Pag. 1 Scheduling In other architectures, buffering and service occur on a per-flow basis That is,
Course of Multimedia Internet (Sub-course Reti Internet Multimediali ), AA 2010-2011 Prof. 4. Scheduling Pag. 1 Scheduling In other architectures, buffering and service occur on a per-flow basis That is,
ARNO Diabete 5. Incidenza, prevalenza e costo del diabete tipo 1
 ARNO Diabete 5 Incidenza, prevalenza e costo del diabete tipo 1 Prof.ssa Graziella Bruno Università di Torino La prof.ssa Bruno dichiara di NON aver ricevuto negli ultimi due anni compensi o finanziamenti
ARNO Diabete 5 Incidenza, prevalenza e costo del diabete tipo 1 Prof.ssa Graziella Bruno Università di Torino La prof.ssa Bruno dichiara di NON aver ricevuto negli ultimi due anni compensi o finanziamenti
Visual communication in hospital
 XI Autism-Europe International Congress 2016 33170 Italy Visual communication in hospital Cinzia Raffin*, Francesco Moscariello^, Laura De Santi^, Roberto Dall Amico^, Marianna Filippini*, Andrea Piai*,
XI Autism-Europe International Congress 2016 33170 Italy Visual communication in hospital Cinzia Raffin*, Francesco Moscariello^, Laura De Santi^, Roberto Dall Amico^, Marianna Filippini*, Andrea Piai*,
Vaccinazioni ed emofilia si/no
 TRAINING CENTER ROMA (Corso malattie emorragiche) 4/8 giugno 2018 Policlinico A. Gemelli (Roma) Vaccinazioni ed emofilia si/no Dott. Matteo Luciani Centro Emostasi e Trombosi Dipartimento Ematologia Oncologia
TRAINING CENTER ROMA (Corso malattie emorragiche) 4/8 giugno 2018 Policlinico A. Gemelli (Roma) Vaccinazioni ed emofilia si/no Dott. Matteo Luciani Centro Emostasi e Trombosi Dipartimento Ematologia Oncologia
TUTTI I TUMORI MALIGNI
 -14 ANNI TUTTI I TUMORI MALIGNI E I NON MALIGNI DEL SISTEMA NERVOSO CENTRALE (SNC) ALL MALIGNANT TUMOURS AND NON-MALIGNANT OF THE CENTRAL NERVOUS SYSTEM (CNS) TUTTI I TUMORI MALIGNI E I NON MALIGNI DELL
-14 ANNI TUTTI I TUMORI MALIGNI E I NON MALIGNI DEL SISTEMA NERVOSO CENTRALE (SNC) ALL MALIGNANT TUMOURS AND NON-MALIGNANT OF THE CENTRAL NERVOUS SYSTEM (CNS) TUTTI I TUMORI MALIGNI E I NON MALIGNI DELL
FACOLTÀ DI MEDICINA VETERINARIA
 FACOLTÀ DI MEDICINA VETERINARIA Corso di Laurea Magistrale in Medicina Veterinaria Tesi di laurea: Valutazione ecografica delle strutture ombelicali del puledro: studio epidemiologico e presentazione di
FACOLTÀ DI MEDICINA VETERINARIA Corso di Laurea Magistrale in Medicina Veterinaria Tesi di laurea: Valutazione ecografica delle strutture ombelicali del puledro: studio epidemiologico e presentazione di
Le dimensioni del problema: epidemiologia e farmaco-economia della BPCO Dr. Claudio Micheletto
 Le dimensioni del problema: epidemiologia e farmaco-economia della BPCO Dr. Claudio Micheletto UOC di Pneumologia Ospedale Mater Salutis Legnago (VR) claudio.micheletto@aulsslegnago.it Soriano JB, Zielinski
Le dimensioni del problema: epidemiologia e farmaco-economia della BPCO Dr. Claudio Micheletto UOC di Pneumologia Ospedale Mater Salutis Legnago (VR) claudio.micheletto@aulsslegnago.it Soriano JB, Zielinski
Inibitori dell enzima DPP4 nell anziano
 XI Edizione PARMA DIABETE Inibitori dell enzima DPP4 nell anziano Francesco Purrello Università di Catania American Diabetes Association Terapia del diabete nel paziente anziano Prevenire la comparsa
XI Edizione PARMA DIABETE Inibitori dell enzima DPP4 nell anziano Francesco Purrello Università di Catania American Diabetes Association Terapia del diabete nel paziente anziano Prevenire la comparsa
Prof. Paolo Nucci Direttore Clinica Oculistica, Università degli Studi di Milano Ospedale San Giuseppe Milano
 MODULO 1 Lezione 1 ESOTROPIA ACCOMODATIVA: OLTRE I CONFINI DEI LIBRI Prof. Paolo Nucci Direttore Clinica Oculistica, Università degli Studi di Milano Ospedale San Giuseppe Milano L ESOTROPIA ACCOMMODATIVA
MODULO 1 Lezione 1 ESOTROPIA ACCOMODATIVA: OLTRE I CONFINI DEI LIBRI Prof. Paolo Nucci Direttore Clinica Oculistica, Università degli Studi di Milano Ospedale San Giuseppe Milano L ESOTROPIA ACCOMMODATIVA
La psicogeriatria: un modello realistico di assistenza all anziano
 Brescia, 23 aprile 2010 Problemi clinici in geriatria La psicogeriatria: un modello realistico di assistenza all anziano Angelo Bianchetti Cosa è la psicogeriatria? Geriatric psychiatry, also known as
Brescia, 23 aprile 2010 Problemi clinici in geriatria La psicogeriatria: un modello realistico di assistenza all anziano Angelo Bianchetti Cosa è la psicogeriatria? Geriatric psychiatry, also known as
Estensioni della genetica mendeliana
 Estensioni della genetica mendeliana Basi molecolari della dominanza e mutazioni Basi molecolari della dominanza e mutazioni Variazioni della dominanza ANIMAZIONE Dominanza incompleta Variazioni della
Estensioni della genetica mendeliana Basi molecolari della dominanza e mutazioni Basi molecolari della dominanza e mutazioni Variazioni della dominanza ANIMAZIONE Dominanza incompleta Variazioni della
Il trattamento a lungo termine (LTT): quali evidenze per quali aspettative
: quali evidenze per quali aspettative") Il trattamento a lungo termine (LTT): quali evidenze per quali aspettative Orazio ZANETTI Società Italiana di Gerontologia e Geriatria U.O. Alzheimer - Centro per la Memoria IRCCS, Centro S.Giovanni di
Il trattamento a lungo termine (LTT): quali evidenze per quali aspettative Orazio ZANETTI Società Italiana di Gerontologia e Geriatria U.O. Alzheimer - Centro per la Memoria IRCCS, Centro S.Giovanni di
EPILESSIA AUTOSOMICA DOMINANTE TEMPORALE LATERALE: aspetti clinici e genetici. Lia Santulli, Federico II, Napoli
 EPILESSIA AUTOSOMICA DOMINANTE TEMPORALE LATERALE: aspetti clinici e genetici Lia Santulli, Federico II, Napoli Ereditarietà autosomica dominante, a penetranza incompleta (67%) Esordio nell adolescenza
EPILESSIA AUTOSOMICA DOMINANTE TEMPORALE LATERALE: aspetti clinici e genetici Lia Santulli, Federico II, Napoli Ereditarietà autosomica dominante, a penetranza incompleta (67%) Esordio nell adolescenza
NASCERE SICURI IL PERCORSO NASCITA. Roma: Commissione Sanità Senato 5 Maggio Giulio Bevilacqua UNIVERSITY OF PARMA - ITALY
 NASCERE SICURI IL PERCORSO NASCITA Roma: Commissione Sanità Senato 5 Maggio 2011 Giulio Bevilacqua UNIVERSITY OF PARMA - ITALY % Sopravvivenza 22-25 eg 35% 30% 25% 20% 15% 10% 5% 22-23eg 24-25eg
NASCERE SICURI IL PERCORSO NASCITA Roma: Commissione Sanità Senato 5 Maggio 2011 Giulio Bevilacqua UNIVERSITY OF PARMA - ITALY % Sopravvivenza 22-25 eg 35% 30% 25% 20% 15% 10% 5% 22-23eg 24-25eg
Sindrome di Sotos: Difetto genetico di base: gene NSD1 e analisi di mutazioni Marina Grasso Laboratorio Genetica Umana E.O.
 Sindrome di Sotos: Difetto genetico di base: gene NSD1 e analisi di mutazioni Marina Grasso Laboratorio Genetica Umana E.O. Ospedali Galliera XVIII Congresso Nazionale Associazione Gulliver Monza 08 marzo
Sindrome di Sotos: Difetto genetico di base: gene NSD1 e analisi di mutazioni Marina Grasso Laboratorio Genetica Umana E.O. Ospedali Galliera XVIII Congresso Nazionale Associazione Gulliver Monza 08 marzo
Scenari per la determinazione del valore di un farmaco oncologico (ASCO ESMO). Perché Ni. Giovanni L. Pappagallo
. Perché Ni. Giovanni L. Pappagallo") Scenari per la determinazione del valore di un farmaco oncologico (ASCO ESMO). Perché Ni Giovanni L. Pappagallo The wide array of treatment options, their attendant clinical impact, and cost to the patient
Scenari per la determinazione del valore di un farmaco oncologico (ASCO ESMO). Perché Ni Giovanni L. Pappagallo The wide array of treatment options, their attendant clinical impact, and cost to the patient
Trattamento della macrodattilia
 1 a GIORNATA NAZIONALE SULLA MACRODATTILIA Monza, 16 dicembre 2017 Trattamento della macrodattilia Studio di fase 1/2 su ARQ 092 in pazienti affetti da malattie da iperaccrescimento e anomalie vascolari
1 a GIORNATA NAZIONALE SULLA MACRODATTILIA Monza, 16 dicembre 2017 Trattamento della macrodattilia Studio di fase 1/2 su ARQ 092 in pazienti affetti da malattie da iperaccrescimento e anomalie vascolari
Feedback and feedforward mechanisms for the control of lower limb posture following anterior cruciate ligament rupture and reconstruction
 DOTTORATO DI RICERCA IN Scienze dello Sport, dell Esercizio Fisico e dell Ergonomia XXVIII CICLO Anni 2012/2013 2014/2015 TITOLO TESI DI DOTTORATO Feedback and feedforward mechanisms for the control of
DOTTORATO DI RICERCA IN Scienze dello Sport, dell Esercizio Fisico e dell Ergonomia XXVIII CICLO Anni 2012/2013 2014/2015 TITOLO TESI DI DOTTORATO Feedback and feedforward mechanisms for the control of
Dott. Stefano Frara Prof. Andrea Giustina Università Vita-Salute, Ospedale San Raffaele Milano
 Dott. Stefano Frara Prof. Andrea Giustina Università Vita-Salute, Ospedale San Raffaele Milano QUANDO IMPIEGARE I FARMACI INIBITORI DEL RIASSORBIMENTO OSSEO? 0 Effetti scheletrici degli inibitori dell
Dott. Stefano Frara Prof. Andrea Giustina Università Vita-Salute, Ospedale San Raffaele Milano QUANDO IMPIEGARE I FARMACI INIBITORI DEL RIASSORBIMENTO OSSEO? 0 Effetti scheletrici degli inibitori dell
La richiesta di informazioni da parte delle Aziende produttrici
 La richiesta di informazioni da parte delle Aziende produttrici Considerazioni personali e spunti di dialogo di Matteo Laurita Longo Lundbeck Italia S.p.A. No info Per vedere l effetto che fa E POI!!!
La richiesta di informazioni da parte delle Aziende produttrici Considerazioni personali e spunti di dialogo di Matteo Laurita Longo Lundbeck Italia S.p.A. No info Per vedere l effetto che fa E POI!!!
Follow-up e programmi di screening: Raccomandazioni europee e questioni aperte. Carlo Senore
 Follow-up e programmi di screening: Raccomandazioni europee e questioni aperte Carlo Senore Abbiamo un approccio standardizzato e condiviso? NO Adenoma a basso rischio Adenoma a rischio intermedio Adenoma
Follow-up e programmi di screening: Raccomandazioni europee e questioni aperte Carlo Senore Abbiamo un approccio standardizzato e condiviso? NO Adenoma a basso rischio Adenoma a rischio intermedio Adenoma
La fatica in neurologia
 La fatica in neurologia M. Pardini DiNOGMI, Università di Genova Organizzazione della presentazione Come definire la fatica (isolata)? Basi neurali della fatica Approccio al paziente con fatica Fatica
La fatica in neurologia M. Pardini DiNOGMI, Università di Genova Organizzazione della presentazione Come definire la fatica (isolata)? Basi neurali della fatica Approccio al paziente con fatica Fatica
Progressi nella vaccinazione estensiva contro la varicella. Aggiornamen2 dalla le4eratura. G. Gabu9 Università degli Studi di Ferrara
 Progressi nella vaccinazione estensiva contro la varicella Aggiornamen2 dalla le4eratura G. Gabu9 Università degli Studi di Ferrara Il contesto Gershon AA, 2007 I quesi2 fondamentali Perché la vaccinazione
Progressi nella vaccinazione estensiva contro la varicella Aggiornamen2 dalla le4eratura G. Gabu9 Università degli Studi di Ferrara Il contesto Gershon AA, 2007 I quesi2 fondamentali Perché la vaccinazione
Questioni di età? Gli aspetti di Anatomia Patologica
 Questioni di età? Gli aspetti di Anatomia Patologica Daniela Fenocchio Citologia e Istologia Diagnostica A.O.U. S. Maria della Misericordia Perugia Una questione di età? Il patologo è un professionista
Questioni di età? Gli aspetti di Anatomia Patologica Daniela Fenocchio Citologia e Istologia Diagnostica A.O.U. S. Maria della Misericordia Perugia Una questione di età? Il patologo è un professionista
LA CLASSIFICAZIONE DELLE OPERAZIONI IN PARTENARIATO PUBBLICO PRIVATO NELL AMBITO DELLE REGOLE EUROPEE
 LA CLASSIFICAZIONE DELLE OPERAZIONI IN PARTENARIATO PUBBLICO PRIVATO NELL AMBITO DELLE REGOLE EUROPEE Gian Paolo Oneto Istat Direzione Centrale della Contabilità Nazionale L ISTAT, attraverso la direzione
LA CLASSIFICAZIONE DELLE OPERAZIONI IN PARTENARIATO PUBBLICO PRIVATO NELL AMBITO DELLE REGOLE EUROPEE Gian Paolo Oneto Istat Direzione Centrale della Contabilità Nazionale L ISTAT, attraverso la direzione
"Può il trattamento osteopatico ridurre il dolore muscolo-scheletrico nei pazienti affetti da malattia di Parkinson?"
 "Può il trattamento osteopatico ridurre il dolore muscolo-scheletrico nei pazienti affetti da malattia di Parkinson?" RIASSUNTO BASI RAZIONALI: il riconoscimento della sintomatologia dolorosa, l identificazione
"Può il trattamento osteopatico ridurre il dolore muscolo-scheletrico nei pazienti affetti da malattia di Parkinson?" RIASSUNTO BASI RAZIONALI: il riconoscimento della sintomatologia dolorosa, l identificazione
AVERE 30 ANNI E VIVERE CON LA MAMMA BIBLIOTECA BIETTI ITALIAN EDITION
 AVERE 30 ANNI E VIVERE CON LA MAMMA BIBLIOTECA BIETTI ITALIAN EDITION READ ONLINE AND DOWNLOAD EBOOK : AVERE 30 ANNI E VIVERE CON LA MAMMA BIBLIOTECA BIETTI ITALIAN EDITION PDF Click button to download
AVERE 30 ANNI E VIVERE CON LA MAMMA BIBLIOTECA BIETTI ITALIAN EDITION READ ONLINE AND DOWNLOAD EBOOK : AVERE 30 ANNI E VIVERE CON LA MAMMA BIBLIOTECA BIETTI ITALIAN EDITION PDF Click button to download
LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI
 Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
Iperpara*roidismo Primario Approccio alla mala3a mul*ghiandolare
 1 CORSO NAZIONALE DI AGGIORNAMENTO IPER[CORSI] AME Iperpara*roidismo Primario Approccio alla mala3a mul*ghiandolare Indagini gene*che: Quando e come? Alfredo Scillitani UO Endocrinologia, Ospedale Casa
1 CORSO NAZIONALE DI AGGIORNAMENTO IPER[CORSI] AME Iperpara*roidismo Primario Approccio alla mala3a mul*ghiandolare Indagini gene*che: Quando e come? Alfredo Scillitani UO Endocrinologia, Ospedale Casa
Ridurre-eliminare la contenzione Ermellina Zanetti (Brescia) E.Zanetti GRG Brescia 1
 E.Zanetti GRG Brescia 1") Ridurre-eliminare la contenzione Ermellina Zanetti (Brescia) E.Zanetti GRG Brescia 1 Premessa e obiettivi Gli infermieri Italiani nel Codice deontologico (Federazione Nazionale dei Collegi IPASVI, 2009)
Ridurre-eliminare la contenzione Ermellina Zanetti (Brescia) E.Zanetti GRG Brescia 1 Premessa e obiettivi Gli infermieri Italiani nel Codice deontologico (Federazione Nazionale dei Collegi IPASVI, 2009)
DIPARTIMENTO : NEURO MOTORIO DIRETTORE DR. ALBERTO FERRARI
 DIPARTIMENTO : NEURO MOTORIO DIRETTORE DR. ALBERTO FERRARI Unità operativa Direttore Referente innovazione e ricerca GERIATRIA ALBERTO FERRARI GIULIO PIOLI MEDICINA FISICA E RIABILITAZIONE NEUROCHIRURGIA
DIPARTIMENTO : NEURO MOTORIO DIRETTORE DR. ALBERTO FERRARI Unità operativa Direttore Referente innovazione e ricerca GERIATRIA ALBERTO FERRARI GIULIO PIOLI MEDICINA FISICA E RIABILITAZIONE NEUROCHIRURGIA
XXIX Congresso Nazionale FCSA Bologna ottobre 2018 STUDIO GIASONE
 XXIX Congresso Nazionale FCSA Bologna 22-23 ottobre 2018 STUDIO GIASONE Prof. Gualtiero Palareti Malattie Cardiovascolari, Università di Bologna Fondazione «Arianna Anticoagulazione», Bologna 2016 In patients
XXIX Congresso Nazionale FCSA Bologna 22-23 ottobre 2018 STUDIO GIASONE Prof. Gualtiero Palareti Malattie Cardiovascolari, Università di Bologna Fondazione «Arianna Anticoagulazione», Bologna 2016 In patients
Combinazioni serie P + MOT
 Combinazioni tra riduttori serie P e MOT Combined series P and MOT reduction units Combinazioni serie P + MOT Sono disponibili varie combinazioni tra precoppie serie P (1 stadio) e riduttori ortogonali
Combinazioni tra riduttori serie P e MOT Combined series P and MOT reduction units Combinazioni serie P + MOT Sono disponibili varie combinazioni tra precoppie serie P (1 stadio) e riduttori ortogonali
PROTOCOLLO SULL UTILIZZO DELLO STIMOLATORE DEL NERVO VAGO COME TERAPIA AGGIUNTIVA NELLE EPILESSIE FARMACORESISTENTI
 PROTOCOLLO SULL UTILIZZO DELLO STIMOLATORE DEL NERVO VAGO COME TERAPIA AGGIUNTIVA NELLE EPILESSIE FARMACORESISTENTI NEL POLICLINICO S. ORSOLA MALPIGHI STATO DATA FIRMA Approvato 20/07/2009 Prof. Emilio
PROTOCOLLO SULL UTILIZZO DELLO STIMOLATORE DEL NERVO VAGO COME TERAPIA AGGIUNTIVA NELLE EPILESSIE FARMACORESISTENTI NEL POLICLINICO S. ORSOLA MALPIGHI STATO DATA FIRMA Approvato 20/07/2009 Prof. Emilio
PET con traccianti per Amiloide
 PET con traccianti per Amiloide Silvia Morbelli UO Medicina Nucleare Ospedale Policlinico San Martino Dipartimento di Scienze della Salute Universita di Genova Articolo su Amyloid PET: come lo avrei voluto
PET con traccianti per Amiloide Silvia Morbelli UO Medicina Nucleare Ospedale Policlinico San Martino Dipartimento di Scienze della Salute Universita di Genova Articolo su Amyloid PET: come lo avrei voluto
Numero delle persone diagnosticate e in cura nel (prevalenza 0.16%)
") Numero delle persone diagnosticate e in cura nel 2012 94.146 (prevalenza 0.16%) Raimondo et al, Notiziario ISS 2014 Numero dei casi di AIDS e incidenza per anno di diagnosi, corretti per ritardo di notifica(1982-2013)
Numero delle persone diagnosticate e in cura nel 2012 94.146 (prevalenza 0.16%) Raimondo et al, Notiziario ISS 2014 Numero dei casi di AIDS e incidenza per anno di diagnosi, corretti per ritardo di notifica(1982-2013)
Prevalenza e trattamento del dismetabolismo nel paziente anziano iperteso: evidenza del mancato controllo dei fattori di rischio cardiovascolare
 Prevalenza e trattamento del dismetabolismo nel paziente anziano iperteso: evidenza del mancato controllo dei fattori di rischio cardiovascolare Sarzani R, Fedecostante M, Spannella F, Dessì-Fulgheri P
Prevalenza e trattamento del dismetabolismo nel paziente anziano iperteso: evidenza del mancato controllo dei fattori di rischio cardiovascolare Sarzani R, Fedecostante M, Spannella F, Dessì-Fulgheri P
CURRICULUM VITAE. Cognome e Nome BIANCHI Amedeo Data di nascita Qualifica
 CURRICULUM VITAE INFORMAZIONI PERSONALI Cognome e Nome BIANCHI Amedeo Data di nascita 06-10-1950 Qualifica Dirigente medico a rapporto esclusivo NEUROLOGIA Incarico attuale Direttore F.F. UOC Neurologia-Neurofisiopatologia
CURRICULUM VITAE INFORMAZIONI PERSONALI Cognome e Nome BIANCHI Amedeo Data di nascita 06-10-1950 Qualifica Dirigente medico a rapporto esclusivo NEUROLOGIA Incarico attuale Direttore F.F. UOC Neurologia-Neurofisiopatologia
Finite Model Theory / Descriptive Complexity: bin
 , CMPSCI 601: Recall From Last Time Lecture 19 Finite Model Theory / Descriptive Compleity: Th: FO L DSPACE Fagin s Th: NP SO. bin is quantifier-free.!#"$&% ('*), 1 Space 0 1 ) % Time $ "$ $ $ "$ $.....
, CMPSCI 601: Recall From Last Time Lecture 19 Finite Model Theory / Descriptive Compleity: Th: FO L DSPACE Fagin s Th: NP SO. bin is quantifier-free.!#"$&% ('*), 1 Space 0 1 ) % Time $ "$ $ $ "$ $.....
Le demenze nella persona molto anziana
 PROGRESSI IN GERIATRIA Brescia Settembre - Dicembre 2012 Le demenze nella persona molto anziana Angelo Bianchetti, 14/09/2012 Angelo Bianchetti Gruppo di Ricerca Geriatrica - Brescia Istituto Clinico S.Anna
PROGRESSI IN GERIATRIA Brescia Settembre - Dicembre 2012 Le demenze nella persona molto anziana Angelo Bianchetti, 14/09/2012 Angelo Bianchetti Gruppo di Ricerca Geriatrica - Brescia Istituto Clinico S.Anna
Dott.ssa Anna Maria Colli U.O. di Cardiologia Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano
 Predictors of medium term treatment of pulmonary hypertension in a population of VLBW and ELBW preterm infants. Dott.ssa Anna Maria Colli U.O. di Cardiologia Fondazione IRCCS Ca' Granda Ospedale Maggiore
Predictors of medium term treatment of pulmonary hypertension in a population of VLBW and ELBW preterm infants. Dott.ssa Anna Maria Colli U.O. di Cardiologia Fondazione IRCCS Ca' Granda Ospedale Maggiore
Valutazione prognostica delle sindromi mielodisplastiche L esperienza genovese
 Valutazione prognostica delle sindromi mielodisplastiche L esperienza genovese Marino Clavio Nicole.a Colombo Raffaella Grasso Fabio Guolo Davide Lovera Maurizio Miglino Paola Mine.o TET2 Muta>ons
Valutazione prognostica delle sindromi mielodisplastiche L esperienza genovese Marino Clavio Nicole.a Colombo Raffaella Grasso Fabio Guolo Davide Lovera Maurizio Miglino Paola Mine.o TET2 Muta>ons
Quale ruolo dei biomarcatori per l anziano con decadimento cognitivo? Patrizia Mecocci
 Quale ruolo dei biomarcatori per l anziano con decadimento cognitivo? Patrizia Mecocci Istituto di Gerontologia e Geriatria Dipartimento di Medicina Università degli Studi di Perugia Alzheimer s pipeline
Quale ruolo dei biomarcatori per l anziano con decadimento cognitivo? Patrizia Mecocci Istituto di Gerontologia e Geriatria Dipartimento di Medicina Università degli Studi di Perugia Alzheimer s pipeline
Epilessia e sindrome epilettica. Epilessia e disturbi cognitivo comportamentali. Epilessia e disturbi cognitivo comportamentali
 Gemma Incorpora Ragusa poggio del sole 3-44 aprile 2009 Epilessia e sindrome epilettica Disturbi del comportamento Iperattività Depressione, ansietà Disturbi di memoria Di apprendimento, di lettura e di
Gemma Incorpora Ragusa poggio del sole 3-44 aprile 2009 Epilessia e sindrome epilettica Disturbi del comportamento Iperattività Depressione, ansietà Disturbi di memoria Di apprendimento, di lettura e di
3DVH. Introduction. Utilizzo di una gamma function 3D (3DVH) e la sua integrazione per tecniche IMRT/Volumetriche
 e la sua integrazione per tecniche IMRT/Volumetriche") Utilizzo di una gamma function 3D (3DVH) e la sua integrazione per tecniche IMRT/Volumetriche Sara Bresciani D.O. Fisica Sanitaria, IRCCS Candiolo sara.bresciani@ircc.it What do these errors mean?? Are
Utilizzo di una gamma function 3D (3DVH) e la sua integrazione per tecniche IMRT/Volumetriche Sara Bresciani D.O. Fisica Sanitaria, IRCCS Candiolo sara.bresciani@ircc.it What do these errors mean?? Are
Malattie autoimmunitarie e gravidanza
 Malattie autoimmunitarie e gravidanza U.O. Cardiologia Pediatrica Osp. Santo Bambino, Catania Discussant: G. Zarbo, M. Vitaliti, G. Pecorella, G. Romeo, S. Nicosia La verità è raramente chiara, mai semplice!
Malattie autoimmunitarie e gravidanza U.O. Cardiologia Pediatrica Osp. Santo Bambino, Catania Discussant: G. Zarbo, M. Vitaliti, G. Pecorella, G. Romeo, S. Nicosia La verità è raramente chiara, mai semplice!
CAPITOLO 2 DEFINIZIONE DELLA DIAGNOSI
 APITOLO 2 DEFINIZIONE DELLA DIAGNOSI 2.1 riteri di definizione dei casi di interesse del ReNaM. Sono inclusi e quindi registrati nel Registro Nazionale dei Mesoteliomi tutti i casi di mesotelioma maligno,
APITOLO 2 DEFINIZIONE DELLA DIAGNOSI 2.1 riteri di definizione dei casi di interesse del ReNaM. Sono inclusi e quindi registrati nel Registro Nazionale dei Mesoteliomi tutti i casi di mesotelioma maligno,
ATTENUATED PSYCHOSIS SYNDROME IN ADOLESCENZA E PREADOLESCENZA: DEFINIZIONE E DIAGNOSI
 1 ATTENUATED PSYCHOSIS SYNDROME IN ADOLESCENZA E PREADOLESCENZA: DEFINIZIONE E DIAGNOSI Marco Armando 1,2 1 Dipartimento di Neuroscienze, UOC di Neuropsichiatria, IRCCS Bambino p, p, Gesù 2 Department
1 ATTENUATED PSYCHOSIS SYNDROME IN ADOLESCENZA E PREADOLESCENZA: DEFINIZIONE E DIAGNOSI Marco Armando 1,2 1 Dipartimento di Neuroscienze, UOC di Neuropsichiatria, IRCCS Bambino p, p, Gesù 2 Department
Cardioversione a tutti i costi? Andrea Bettella Pronto Soccorso e Medicina d Urgenza Ospedale S. Antonio - Padova
 Andrea Bettella Pronto Soccorso e Medicina d Urgenza Ospedale S. Antonio - Padova ? Archives of Cardiovascular Disease (2012) 105, 226 238 Archives of Cardiovascular Disease (2012) 105, 226 238 Archives
Andrea Bettella Pronto Soccorso e Medicina d Urgenza Ospedale S. Antonio - Padova ? Archives of Cardiovascular Disease (2012) 105, 226 238 Archives of Cardiovascular Disease (2012) 105, 226 238 Archives
Management della Toxoplasmosi in gravidanza
 Management della Toxoplasmosi in gravidanza Cenni sul protozoo Oocisti,trofozoite,bradizoite,cisti Fisiopatologia Infezione acuta : parassitemica Infezione cronica: senza parassitemia Clinica (dell infezione
Management della Toxoplasmosi in gravidanza Cenni sul protozoo Oocisti,trofozoite,bradizoite,cisti Fisiopatologia Infezione acuta : parassitemica Infezione cronica: senza parassitemia Clinica (dell infezione
Università degli Studi di Napoli Federico II
 Università degli Studi di Napoli Federico II Dipartimento di Ingegneria Civile, Edile e Ambientale Relatore: Classe delle lauree in Ingegneria Civile e Ambientale, Classe n 35. Corso di Laurea Magistrale
Università degli Studi di Napoli Federico II Dipartimento di Ingegneria Civile, Edile e Ambientale Relatore: Classe delle lauree in Ingegneria Civile e Ambientale, Classe n 35. Corso di Laurea Magistrale
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali. Silvia Sansavini
 CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
Valutazione delle evidenze: studio IBIS II
 Valutazione delle evidenze: studio IBIS II Marta Pestrin Sandro Pitigliani Medical Oncology Dept. Hospital of Prato Istituto Toscano Tumori, Prato, Italy Hormonal strategies for breast cancer shown to
Valutazione delle evidenze: studio IBIS II Marta Pestrin Sandro Pitigliani Medical Oncology Dept. Hospital of Prato Istituto Toscano Tumori, Prato, Italy Hormonal strategies for breast cancer shown to
DI.S.SAL Dipartimento di Scienze della Salute Università degli Studi di Genova Vaccinazioni in Italia: obiettivi raggiunti e strategie per il futuro
 DI.S.SAL Dipartimento di Scienze della Salute Università degli Studi di Genova Vaccinazioni in Italia: obiettivi raggiunti e strategie per il futuro PIANO SANITARIO NAZIONALE 2003-2005 La recente disponibilità
DI.S.SAL Dipartimento di Scienze della Salute Università degli Studi di Genova Vaccinazioni in Italia: obiettivi raggiunti e strategie per il futuro PIANO SANITARIO NAZIONALE 2003-2005 La recente disponibilità
Misure riassuntive di effetto per varie tipologie di variabili statistiche
 Misure riassuntive di effetto per varie tipologie di variabili statistiche VARIABILE DI RISPOSTA di tipo qualitativo esprime categorie di risposta del tipo successo / insuccesso (di un trattamento somministrato).
Misure riassuntive di effetto per varie tipologie di variabili statistiche VARIABILE DI RISPOSTA di tipo qualitativo esprime categorie di risposta del tipo successo / insuccesso (di un trattamento somministrato).
NATIONAL SPORT SCHOOL
 NATIONAL SPORT SCHOOL Mark HALF-YEARLY EXAMINATION 2016 Level 4-6 FORM 1 ITALIAN TIME: 30 minutes LISTENING COMPREHENSION TEST (20 punti) Teacher s Paper Please first read the instructions carefully by
NATIONAL SPORT SCHOOL Mark HALF-YEARLY EXAMINATION 2016 Level 4-6 FORM 1 ITALIAN TIME: 30 minutes LISTENING COMPREHENSION TEST (20 punti) Teacher s Paper Please first read the instructions carefully by
DISCLOSURE INFORMATION
 DISCLOSURE INFORMATION Prof. Antonio Palla negli ultimi due anni ho avuto i seguenti rapporti anche di finanziamento con soggetti portatori di interessi commerciali in campo sanitario: NESSUNO L imaging
DISCLOSURE INFORMATION Prof. Antonio Palla negli ultimi due anni ho avuto i seguenti rapporti anche di finanziamento con soggetti portatori di interessi commerciali in campo sanitario: NESSUNO L imaging
Corrado Amato U.O.S.D. di Angiologia Università di Palermo
 EE Corrado Amato U.O.S.D. di Angiologia Università di Palermo Nuove informazioni sull epidemiologia delle AOP: -Prevalenza ed incidenza -Differenze di razza -Differenza di genere -Patologia dei paesi
EE Corrado Amato U.O.S.D. di Angiologia Università di Palermo Nuove informazioni sull epidemiologia delle AOP: -Prevalenza ed incidenza -Differenze di razza -Differenza di genere -Patologia dei paesi
Si faccia riferimento all Allegato A - OPS 2016, problema ricorrente REGOLE E DEDUZIONI, pagina 2.
 Scuola Sec. SECONDO Grado Gara 2 IND - 15/16 ESERCIZIO 1 Si faccia riferimento all Allegato A - OPS 2016, problema ricorrente REGOLE E DEDUZIONI, pagina 2. Sono date le seguenti regole: regola(1,[a],b)
Scuola Sec. SECONDO Grado Gara 2 IND - 15/16 ESERCIZIO 1 Si faccia riferimento all Allegato A - OPS 2016, problema ricorrente REGOLE E DEDUZIONI, pagina 2. Sono date le seguenti regole: regola(1,[a],b)
La questione dei Multiple Comparisons
 La questione dei Multiple Comparisons Massimo Borelli May 7, 2014 Massimo Borelli () La questione dei Multiple Comparisons May 7, 2014 1 / 27 Contenuti 1 Un errore tanto grave quanto frequente 2 i vantaggi
La questione dei Multiple Comparisons Massimo Borelli May 7, 2014 Massimo Borelli () La questione dei Multiple Comparisons May 7, 2014 1 / 27 Contenuti 1 Un errore tanto grave quanto frequente 2 i vantaggi
Vai al contenuto multimediale
 A06 Vai al contenuto multimediale Maria Antonietta Lepore Ruolo della procalcitonina e della proteina C reattiva nelle infezioni batteriche Aracne editrice www.aracneeditrice.it info@aracneeditrice.it
A06 Vai al contenuto multimediale Maria Antonietta Lepore Ruolo della procalcitonina e della proteina C reattiva nelle infezioni batteriche Aracne editrice www.aracneeditrice.it info@aracneeditrice.it
QUAL E IL RUOLO DELL ABLAZIONE DI FA NELLA GESTIONE DEL PAZIENTE A RISCHIO DI ICTUS
 Ospedale SS Antonio e Biagio e Cesare Arrigo QUAL E IL RUOLO DELL ABLAZIONE DI FA NELLA GESTIONE DEL PAZIENTE A RISCHIO DI ICTUS Alice Scopinaro MD, Phd 1 2 3 5 DIAGNOSI DI FIBRILLAZIONE ATRIALE STRATIFICAZIONE
Ospedale SS Antonio e Biagio e Cesare Arrigo QUAL E IL RUOLO DELL ABLAZIONE DI FA NELLA GESTIONE DEL PAZIENTE A RISCHIO DI ICTUS Alice Scopinaro MD, Phd 1 2 3 5 DIAGNOSI DI FIBRILLAZIONE ATRIALE STRATIFICAZIONE
Cosa abbiamo imparato dagli studi SUP ed EGSYS-follow-up? Andrea Ungar. Syncope Unit
 Cosa abbiamo imparato dagli studi SUP ed EGSYS-follow-up? Andrea Ungar Syncope Unit Cardiologia e Medicina Geriatrica Dipartimento del Cuore e dei Vasi Azienda Ospedaliero-Universitaria Careggi - Firenze
Cosa abbiamo imparato dagli studi SUP ed EGSYS-follow-up? Andrea Ungar Syncope Unit Cardiologia e Medicina Geriatrica Dipartimento del Cuore e dei Vasi Azienda Ospedaliero-Universitaria Careggi - Firenze
Gestione del trauma cranico pediatrico
 Gestione del trauma cranico pediatrico Forze traumatiche del Danno Primario: impatto lesioni cerebrali dirette lesioni da contraccolpo 15 Forze traumatiche del Danno Primario: accelerazione/decelarazione,
Gestione del trauma cranico pediatrico Forze traumatiche del Danno Primario: impatto lesioni cerebrali dirette lesioni da contraccolpo 15 Forze traumatiche del Danno Primario: accelerazione/decelarazione,
LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI
 Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
Read Online and Download Ebook LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO TESTAMENTO VERSIONE RIVEDUTA BY GIOVANNI LUZZI DOWNLOAD EBOOK : LA SACRA BIBBIA: OSSIA L'ANTICO E IL NUOVO Click link bellow and
MYC nei Linfomi B Diffusi a Grandi Cellule
 Padova, 9 novembre 2012 MYC nei Linfomi B Diffusi a Grandi Cellule Analisi FISH e immunoistochimica Fabio Facchetti Anatomia Patologica Spedali Civili-Università Brescia Kramer, Blood 1998 Macpherson,
Padova, 9 novembre 2012 MYC nei Linfomi B Diffusi a Grandi Cellule Analisi FISH e immunoistochimica Fabio Facchetti Anatomia Patologica Spedali Civili-Università Brescia Kramer, Blood 1998 Macpherson,
Il percorso del paziente dopo il DEA. Dal documento EHRA sulla Syncope Unit al mondo reale
 Il percorso del paziente dopo il DEA. Dal documento EHRA sulla Syncope Unit al mondo reale Martina Rafanelli Syncope Unit, Geriatria e UTIG, Università degli Studi di Firenze, AOU Careggi Firenze Syncopeis
Il percorso del paziente dopo il DEA. Dal documento EHRA sulla Syncope Unit al mondo reale Martina Rafanelli Syncope Unit, Geriatria e UTIG, Università degli Studi di Firenze, AOU Careggi Firenze Syncopeis