Odontotecnico Fabbricante di Dispositivi medici su misura. responsabilità adempimenti prospettive. Seminario SNO CNA Veneto 29 ottobre 2015
|
|
|
- Baldassare Silvestri
- 7 anni fa
- Visualizzazioni
Transcript
1 Santa Apollonia Odontotecnico Fabbricante di Dispositivi medici su misura responsabilità adempimenti prospettive Seminario SNO CNA Veneto 29 ottobre 2015 Relatore: Sandro Storelli
2 Norme di riferimento
3 Profilo professionale
4 Accesso alla professione & Requisiti per l impresa
5 Odontotecnico in Europa

6 Dispositivi medici su misura
7 Aspetti ambientali - VENETO Adempimenti emissioni in atmosfera scarichi idrici rifiuti impatto acustico Le emissioni da attività di odontotecnico sono classificate come emissioni poco significative. Devono essere convogliate e non è previsto il rilascio di un autorizzazione. Le acque reflue provenienti dall attività di odontotecnico sono considerate assimilabili alle acque reflue domestiche e non richiedono un autorizzazione specifica. E comunque necessario verificare il regolamento dell ente gestore delle fognature per le modalità di rilascio dell autorizzazione allo scarico in fognatura. L attività di odontotecnico genera numerose tipologie di rifiuti classificati come pericolosi e non pericolosi (rivestimenti a base di gesso, la pomice, le resine, le cere, rivestimenti e materiali refrattari, ecc.). Per i rifiuti non pericolosi il comune può aver disposto l assimilabilità ai rifiuti urbani con la conseguente possibilità per i produttori di conferirli al servizio pubblico. Per i rifiuti pericolosi e per quelli non assimilati agli urbani il produttore deve osservare la normativa specifica in merito al deposito temporaneo e alla gestione amministrativa (registro di carico e scarico, formulario per il trasporto, denuncia annuale e SISTRI). L attività è considerata a bassa rumorosità ed è esonerata dall obbligo di presentazione della documentazione per la valutazione di impatto acustico. Può essere comunque dovuta una dichiarazione sostitutiva di atto di notorietà attestante il rispetto dei limiti stabiliti nella zonizzazione acustica del territorio comunale. Riferimenti normativi D.Lgs. 152/2006, art. 272, commi 1 e 5. D.Lgs. 152/2006, artt DPR 19 ottobre 2011 n 227. DCR Veneto n. 107 del 05/11/2009 D.Lgs. 152/2006, artt. 183, 188 ter, 189, 190 e 193. Legge 25 gennaio 1994, n. 70 DPR n.227 del 19/10/2011 art. 4 L. 26/10/1995 n. 447 art. 8, commi 2, 3 e 4
8 Adempimenti ambiente & sicurezza sul lavoro Controlli impianti verifica periodica della messa a terra controllo periodico dell'impianto elettrico verifica semestrale degli estintori verifica periodica della caldaia a gas Sorveglianza sanitaria da parte di un medico competente del lavoro (visite mediche, sopralluogo in azienda, analisi etc...) Formazione Sicurezza sul lavoro Rspp (48 ore - datore di lavoro) primo soccorso (12 ore) prevenzione incendi (8 ore) formazione base lavoratori 4 ore formazione specifica lavoratori 12 ore (alto rischio - ateco 2007: ) Valutazioni rischi documento valutazione rischi valutazione rischio vibrazioni meccaniche mano braccio (micromotori) valutazione rischio rumore valutazione rischio chimico valutazione rischio stress lavoro correlato valutazione rischio lavoratrici madri

9 Innovazione ed evoluzione L evoluzione è continua, in particolare in ambito medicale In media, il ciclo di vita media dei prodotti non supera i 5 anni.

10 Complessità, correlazioni e variabili del mercato
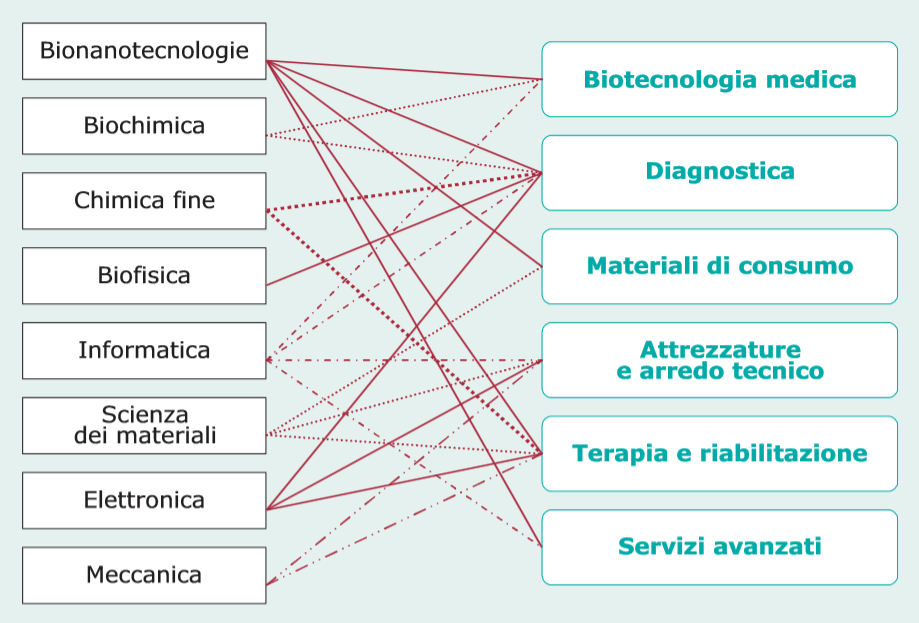
11 Dipendenze tecnologiche

12 Dentale: fatturato in milioni Euro a prezzi ex fabbrica Italia Produzione: 1200 milioni Euro Addetti Posizionamento: Terza al mondo

13 Dentale: filiera produttiva
14 Dentale: posizionamento famiglie prodotto e attrattività
15 Dalla 93/42 alla gestione del rischio

16 Processo controllato e buone prassi

17 Evoluzione gestione del rischio


18 Aspettative e ciclo di vita
19 REGOLAMENTO DM Entrerà in vigore gradualmente nel periodo Il Regolamento sarà direttamente applicabiledagli Stati Membri.

20 Quadro normativo per i dispositivi medici Direttiva 90/385/CEE del 20 giugno 1990 concernente i dispositivi medici impiantabili attivi Recepita in Italia con Decreto Legislativo n.507/1992 Direttiva 93/42/CEE del 14 giugno 1993 concernente i dispositivi medici Recepita in Italia con Decreto Legislativo n.46/97 Emendate (non sostituite) dalla Direttiva 2007/47/CE Recepita in Italia con Decreto Legislativo n.37/2010 Regolamento Europeo applicazione prevista: 2017
21 NUOVI ADEMPIMENTI PER I FABBRICANTI Identificare i fornitori e i clienti dei dispositivi medici Tracciabilità dei DM Persona qualificata all interno dell azienda Requisito di un Responsabile per la conformità normativa e la parte regolatoria avente determinate qualifiche Adattare ai DM un Codice Identificativo Unico (UDI) Europeo UDI Rietichettatura e/o riconfezionamento Stabilite condizioni più chiare Fabbricanti, mandatari e importatori devono registrarsi e registrare i dispositivi Banca dati Europea centrale Ricondizionamento monouso Norme rigorose e più chiare Rendere disponibile al pubblico gli elementi chiave di supporto ai dati clinici Sintesi prestazioni di sicurezza per DM ad alto rischio Indicazioni su Dispositivi impiantabili Maggior enfasi sul fornire ai pazienti avvertenze e informazioni
22 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENTO TESTO CONSIDERAZIONI Art. 1 Definizioni, campo di applicazione «dispositivo medico»: qualunque strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compreso il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo, destinato dal fabbricante ad essere impiegato sull uomo a fini di: diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia; diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap; studio, sostituzione o modifica dell'anatomia o di un processo fisiologico; intervento sul concepimento, la cui azione principale voluta nel o sul corpo umano no sia conseguita con mezzi farmacologici né immunologici né mediante metabolismo, ma la cui funzione possa essere assistita da questi mezzi; Art. 2 - Definizioni "dispositivo medico": qualunque strumento, apparecchio, apparecchiatura, software, impianto, reagente, materiale o altro articolo, destinato dal fabbricante ad essere impiegato sull'uomo, da solo o in combinazione, per una o più delle seguenti finalità mediche specifiche:diagnosi, prevenzione, monitoraggio, trattamento o attenuazione di malattie, diagnosi, monitoraggio, trattamento, attenuazione o compensazione di una lesione o di una disabilità, studio, sostituzione o modifica dell'anatomia oppure di un processo o stato fisiologico, controllo del concepimento o supporto al concepimento, disinfezione o sterilizzazione di qualsiasi prodotto citato sopra, che non esercita nel o sul corpo umano l'azione principale cui è destinato mediante mezzi farmacologici, immunologici o metabolici, ma la cui funzione può essere coadiuvata da tali mezzi. I dispositivi impiantabili o altri prodotti invasivi destinati ad essere utilizzati per l'uomo, elencati nell'allegato XV, sono considerati dispositivi medici, indipendentemente dal fatto che siano destinati o meno dal fabbricante ad essere impiegati a scopo medico; Viene ampliata la definizione di dispositivo medico. Nel nuovo regolamento rientrano tra i dispositivi medici, gli strumenti di disinfezione o sterilizzazione di dispositivi medici, dispositivi impiantabili o invasivi destinati per l uomo ed elencati nell allegato XV anche quando non destinati a scopo medici. (es. lenti a contatto non correttive o protesi a scopo estetico) campo applicativo
23 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENTO TESTO CONSIDERAZIONI Art. 1 definizioni, campo di applicazione dispositivo su misura: qualsiasi dispositivo fabbricato appositamente sulla base della prescrizione scritta di un medico debitamente qualificato e indicante, sotto la responsabilità del medesimo, le caratteristiche di progettazione e destinato ad essere utilizzato solo per un determinato paziente. La prescrizione di cui sopra può essere redatta anche da un'altra persona la quale vi sia autorizzata in virtù delle sue qualificazioni professionali. I dispositivi fabbricati con metodi di fabbricazione continua o in serie che devono essere successivamente adattati, per soddisfare un'esigenza specifica del medico o di un altro utilizzatore professionale, non sono considerati dispositivi su misura; Art. 2 definizioni "dispositivo su misura": qualsiasi dispositivo fabbricato appositamente sulla base della prescrizione scritta di un medico, di un dentista o di qualsiasi altra persona autorizzata dalla legislazione nazionale in virtù delle sue qualifiche professionali, che precisi, sotto la propria responsabilità, le caratteristiche specifiche di progettazione, e che è destinato ad essere utilizzato solo per un determinato paziente. I dispositivi fabbricati in serie che devono essere adattati per soddisfare le esigenze specifiche di un medico, un dentista o un altro utilizzatore professionale e i dispositivi che sono fabbricati in serie mediante processi di fabbricazione industriale conformemente alle prescrizioni scritte di medici, dentisti o qualsiasi altra persona autorizzata non sono tuttavia considerati dispositivi su misura; Attualmente ha facoltà di prescrivere un dispositivo medico su misura un medico qualificato o un altra persona autorizzata in virtù delle sue qualifiche professionali. Nel nuovo regolamento viene meglio dettagliato questo aspetto e viene specificato che la prescrizione può essere scritta da un medico, un dentista o qualsiasi altra persona autorizzata dalla legislazione nazionale in virtù delle sue qualifiche professionali. prescrittore
24 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENTO TESTO CONSIDERAZIONI Art. 1 definizioni, campo di applicazione fabbricante: la persona fisica o giuridica responsabile della progettazione, della fabbricazione, dell'imballaggio e dell'etichettatura di un dispositivo in vista dell'immissione in commercio a proprio nome, indipendentemente dal fatto che queste operazioni siano eseguite da questa stessa persona o da un terzo per suo conto. Gli obblighi della presente direttiva che si impongono al fabbricante valgono anche per la persona fisica o giuridica che compone, provvede all'imballaggio, tratta, rimette a nuovo e/o etichetta uno o più prodotti prefabbricati e/o assegna loro la destinazione di dispositivo in vista dell'immissione in commercio a proprio nome. Il presente comma non si applica alla persona la quale, senza essere il fabbricante ai sensi del primo comma compone o adatta dispositivi, già immessi in commercio in funzione della loro destinazione ad un singolo paziente; Art. 2 definizioni "fabbricante": la persona fisica o giuridica che fabbrica o rimette a nuovo un dispositivo oppure lo fa progettare, fabbricare o rimettere a nuovo, e lo commercializza apponendovi il suo nome o marchio. Ai fini della definizione di fabbricante, per rimessa a nuovo si intende la ricostruzione completa di un dispositivo già immesso sul mercato o messo in servizio, o la fabbricazione di un nuovo dispositivo a partire da dispositivi usati, per renderlo conforme al presente regolamento, unitamente al conferimento di un nuovo periodo di vita utile al dispositivo rimesso a nuovo; Nel nuovo regolamento viene spiegato il concetto di rimessa a nuovo di un dispositivo. rimessa a nuovo
25 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENTO TESTO CONSIDERAZIONI Art. 2 - Immissione in commercio e messa in servizio Gli Stati membri adottano le disposizioni necessarie affinché i dispostivi possano essere immessi in commercio e/o messi in servizio unicamente qualora rispondano alle condizioni prescritte dalla presente direttiva, siano correttamente forniti e installati, siano oggetto di un'adeguata manutenzione e siano utilizzati in conformità della loro destinazione. Art. 4 - Immissione sul mercato e messa in servizio Un dispositivo può essere immesso sul mercato o messo in servizio solo se è conforme al presente regolamento qualora sia debitamente fornito e correttamente installato, oggetto di un'adeguata manutenzione e utilizzato conformemente alla sua destinazione. 2. Un dispositivo soddisfa i requisiti generali di sicurezza e prestazione ad esso applicabili, tenuto conto della sua destinazione. I requisiti generali di sicurezza e prestazione sono enunciati nell'allegato I. 3. La dimostrazione della conformità ai requisiti generali di sicurezza e prestazione comprende una valutazione clinica a norma dell'articolo I dispositivi fabbricati e utilizzati in una singola istituzione sanitaria sono considerati messi in servizio. Le disposizioni in materia di marcatura CE di cui all'articolo 18 e gli obblighi di cui agli articoli da 23 a 27 non si applicano a tali dispositivi, purché essi siano fabbricati e utilizzati nel quadro del sistema unico di gestione della qualità dell'istituzione sanitaria. 5. Alla Commissione è conferito il potere di adottare atti delegati conformemente all'articolo 89 per modificare o integrare, alla luce del progresso tecnico e tenuto conto degli utilizzatori o dei pazienti cui sono destinati i dispositivi, i requisiti generali di sicurezza e prestazione enunciati nell'allegato I, comprese le informazioni fornite dal fabbricante. L articolo viene ampliato e si specifica già nel presente che il dispositivo può essere immesso sul mercato se soddisfa i requisiti generali di sicurezza e prestazioni che comprendono una valutazione clinica. Sono considerati messi in servizio i dispositivi fabbricati e utilizzati in una singola istituzione sanitaria. In ogni caso per tali dispositivi non è necessaria la marcatura CE e l inserimento nella banca dati europea, purché utilizzati nel quadro del sistema unico di gestione della qualità dell istituzione sanitaria. valutazione clinica
26 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENT O - - Art. 13 Persona responsabile del rispetto della normativa TESTO I fabbricanti, all'interno della loro organizzazione, dispongono di almeno una persona qualificata in possesso di conoscenze specializzate nel settore dei dispositivi medici.. Il presente paragrafo non si applica ai fabbricanti di dispositivi su misura che sono microimprese quali definite nella raccomandazione 2003/361/CE della Commissione. CONSIDERAZIONI Nel nuovo regolamento è stato introdotto il concetto di persona responsabile in ambito regolatorio. I fabbricanti di dispositivi medici devono disporre di tale figura all interno della propria organizzazione. Per i fabbricanti di dispositivi medici su misura questo articolo non si applica per aziende appartenenti alle microimprese. ambito regolatorio
27 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. TESTO Art. di riferimento NUOVA PROPOSTA DI REGOLAMENT O - - Art Dispositivi per destinazioni particolari TESTO I dispositivi su misura sono muniti della dichiarazione di cui all'allegato XI, che è messa a disposizione di un determinato paziente o utilizzatore, identificato mediante il nome, un acronimo o un codice numerico. Gli Stati membri possono stabilire che il fabbricante di un dispositivo su misura debba presentare all'autorità competente un elenco dei dispositivi di questo tipo messi a disposizione nel loro territorio. CONSIDERAZIONI I dispositivi medici su misura si dichiarano conformi al nuovo regolamento se soddisfano i requisiti dell allegato XI (non più VIII). Gli stati membri possono stabilire regole di registrazione di tali dispositivi immessi in commercio. Allegato XI
28 ART. DI RIFERIMENTO Dir. 93/42/CEE e s.m.i. Art. 10 Informazioni riguardanti incidenti verificatesi dopo l immissione in commercio TESTO 1. Gli Stati membri prendono i provvedimenti necessari affinché i dati loro comunicati secondo il disposto della presente direttiva e riguardanti gli incidenti di seguito elencati che hanno coinvolto un dispositivo appartenente ad una delle classi I, IIa, IIb o III siano classificati e valutati a livello centrale: a) qualsiasi disfunzione o deterioramento delle caratteristiche e/o delle prestazioni nonché qualsiasi carenza nell'etichettatura o nelle istruzioni per l'uso di un dispositivo che possano causare o abbiano causato la morte o un grave peggioramento dello stato di salute del paziente o di un utilizzatore; b) qualsiasi motivo di ordine tecnico o sanitario connesso alle caratteristiche o alle prestazioni di un dispositivo per i motivi di cui alla lettera a), che abbia causato il ritiro sistematico dal mercato da parte del fabbricante dei dispositivi appartenenti allo stesso tipo. 2. Se prescrivono ai medici o agli organismi sanitari di comunicare gli incidenti contemplati al paragrafo 1 alle autorità competenti, gli Stati membri adottano le misure necessarie affinché il fabbricante del dispositivo in questione oppure il suo mandatario ne sia informato. 3. Dopo aver effettuato una valutazione, se possibile insieme al fabbricante o al suo mandatario, gli Stati membri, fatto salvo l articolo 8, informano immediatamente la Commissione e gli altri Stati membri in merito alle misure che sono state adottate o che sono previste per ridurre al minimo il verificarsi degli incidenti di cui al paragrafo 1, ivi incluse informazioni sugli incidenti alla base. 4. Le misure atte all adozione di procedure per l attuazione del presente articolo sono adottate secondo la procedura di regolamentazione di cui all articolo 7, paragrafo 2 Art. di riferimento NUOVA PROPOSTA DI REGOLAMENT O Art. 61 Segnalazioni di incidenti e azioni correttive di sicurezza TESTO I fabbricanti di dispositivi diversi dai dispositivi su misura e da quelli oggetto di indagine segnalano tramite il sistema elettronico di cui all'articolo 62: (a) qualsiasi incidente grave relativo a dispositivi messi a disposizione sul mercato dell'unione; (b) qualsiasi azione correttiva di sicurezza relativa a dispositivi messi a disposizione sul mercato dell'unione, incluse le azioni correttive di sicurezza intraprese in un paese terzo in relazione a un dispositivo messo legittimamente a disposizione anche sul mercato dell'unione se l'azione correttiva in questione non è causata solo dal dispositivo messo a disposizione nel paese terzo. I fabbricanti trasmettono la segnalazione di cui al primo comma quanto prima, e comunque non oltre 15 giorni dopo aver avuto conoscenza dell'evento e del nesso causale, anche solo ragionevolmente possibile, con il loro dispositivo. Il termine per le segnalazioni è commisurato alla gravità dell'incidente. Per assicurare segnalazioni tempestive, il fabbricante può, se del caso, presentare una relazione iniziale incompleta, seguita da una relazione completa. 2. Per incidenti gravi simili che si verificano con lo stesso dispositivo o tipo di dispositivo e dei quali è stata individuata la causa principale o che sono stati oggetto di un'azione correttiva di sicurezza, i fabbricanti possono presentare relazioni di sintesi periodiche anziché singole relazioni sugli incidenti, purché le autorità competenti di cui all'articolo 62, paragrafo 5, lettere a), b) e c), abbiano convenuto con il fabbricante il formato, il contenuto e la frequenza delle relazioni di sintesi periodiche. 3. Gli Stati membri adottano tutte le misure atte ad incoraggiare gli operatori sanitari, gli utilizzatori e i pazienti a segnalare alle loro autorità competenti gli incidenti gravi sospetti di cui al paragrafo 1, lettera a). Tali segnalazioni sono registrate in modo centralizzato a livello nazionale. Quando un'autorità competente di uno Stato membro riceve tali segnalazioni, adotta le misure necessarie affinché il fabbricante del dispositivo in questione sia informato dell'incidente. Il fabbricante provvede a un follow-up adeguato. Gli Stati membri coordinano tra loro l'elaborazione di moduli standard online strutturati per la segnalazione di incidenti gravi da parte di operatori sanitari, pazienti e utilizzatori. 4. I fabbricanti di dispositivi su misura segnalano eventuali incidenti gravi e azioni correttive di sicurezza di cui al paragrafo 1 all'autorità competente dello Stato membro nel quale il dispositivo in questione è stato messo a disposizione. CONSIDERAZIONI Il regolamento richiede di segnalare attraverso sistema elettronico eventuali incidenti, azioni correttive definite sul prodotto. Rimane l obbligo per i fabbricanti di dispositivi medici su misura di informare all autorità competente gravi incidenti avvenuti durante l utilizzo del dispositivo. incidenti

29 Registrazione fabbricanti

30 ITCA
31

32 Sistema di vigilanza
33 Modulo segnalazione
34 Fascicolo Tecnico Prescrizione

35 Fascicolo Tecnico Dichiarazione conformità
36 Fascicolo Tecnico Classificazione

37 Fascicolo Tecnico specifiche dei materiali
38 Fascicolo Tecnico Flusso processo
39 Fascicolo Tecnico Controllo processo
40 Fascicolo Tecnico Progetto tecnico
41 Fascicolo Tecnico Verifiche
42 Fascicolo Tecnico Gestione non conformità
43 Fascicolo Tecnico Lavorazioni esterne
44 Fascicolo Tecnico del Prototipo unico
45 Fascicolo Tecnico RES
46 Fascicolo Tecnico Analisi dei rischi
47 Fascicolo Tecnico Pericoli & rischi
48 Fascicolo Tecnico Informazioni e istruzioni - Etichetta
49 Qualità e termini di garanzia Quanto dura la garanzia? Per il fabbricante, un primo passaggio è la classificazione del dispositivo. Serve per individuarne la classe, ma anche la durata. I DM su misura", a seconda della loro destinazione d'uso, possono avere diversa durata di vita. Si presume ad esempio che un dispositivo provvisorio abbia una prevedibile durata di vita di 30 giorni, mentre una protesi definitiva può ragionevolmente durare cinque anni. La durabilità di un dispositivo varia in funzione di fattori diversi. Nelle evidenze della pratica ed esperienza professionale, si trovano i riferimenti utili. Le caratteristiche di qualità derivano in particolare da materiali utilizzati, processi di lavorazione controllati, prove e test applicati. Differenza tra garanzia legale e convenzionale Le garanzie convenzionali non sostituiscono né limitano quella legale di conformità, rispetto alla quale possono avere invece diversa ampiezza e durata. Le garanzie convenzionali sono diverse e aggiuntive rispetto alla garanzia legale di conformità. Nel caso del dispositivo medico su misura, la garanzia convenzionale può prevedere ampiezza e/o durata, coerenti con le caratteristiche di qualità e con il valore attribuito allo specifico dispositivo.
50 Fatturazione diretta all utilizzatore finale Va premesso che la professione di odontotecnico è considerata arte ausiliaria dal R.D. 1334/1928, che, all art. 11, prevede espressamente che Gli odontotecnici sono autorizzati unicamente a costruire apparecchi di protesi dentaria su modelli tratti dalle impronte loro fornite dai medici chirurghi e dagli abilitati a norma di legge all'esercizio della odontoiatria e protesi dentaria, con le indicazioni del tipo di protesi È in ogni caso vietato agli odontotecnici di esercitare, anche alla presenza ed in concorso del medico o dell'abilitato all'odontoiatria, alcuna manovra, cruenta o incruenta, nella bocca del paziente, sana o ammalata. Il divieto, quindi, di operare direttamente nei confronti del paziente è limitato alle operazioni svolte nella bocca del paziente. Nulla dice la norma in merito alla possibilità per l odontotecnico di fatturare direttamente al cliente la prestazione eseguita. Probabilmente, il dubbio che la fatturazione diretta sia interdetta all odontotecnico, è sorto in quanto il Decreto del 1928 non la prevede espressamente, a differenza di quanto ad esempio prescritto per gli ottici. Interpellata sul punto, l Agenzia delle Entrate non ha mai avuto dubbi: essendo la figura dell odontotecnico inserita tra le professioni ausiliarie sanitarie, non solo vi è titolo per l emissione diretta della fattura, ma essa, se accompagnata dalla prescrizione medica, può essere detratta dal cliente, al pari di ogni altra prestazione sanitaria (Circolare 39/E del ). Peraltro, già con la nota del 2 maggio 1995 il Ministero delle Finanze aveva chiarito che le prestazioni rese dagli odontotecnici vanno esentate dall IVA essendo rese da esercenti le professioni sanitarie soggette a vigilanza. Questa nota richiamava il parere del Ministero della Salute del , secondo il quale le prestazioni di riparazione delle protesi, anche se eseguite in assenza di una prescrizione medica e anche se fatturate direttamente al paziente, sono in ogni caso funzionalmente connesse all attività sanitaria. Di fatto, viene così ammesso, dal Ministero della Salute, la possibilità di un rapporto diretto tra odontotecnico e cliente. Si tratta certo di un ammissione parziale, riferita solo alla riparazione delle protesi, ma vale a ribadire il principio per il quale il divieto di interazione con il paziente riguarda, per l odontotecnico, solo le manovre nella bocca del paziente, non certo il rapporto di clientela. Nel 2012, a seguito della pubblicazione sul Corriere della Sera di una nota di precisazione dell Agenzia delle Entrate, che riteneva non necessaria la prescrizione medica per la detraibilità delle fatture emesse direttamente dall odontotecnico al paziente, si è accesa una polemica del CAO, che, partendo dalla necessità della prescrizione medica, come presupposto della prestazione dell odontotecnico, riteneva che il divieto di rapporto diretto con la clientela comportasse, necessariamente, anche il divieto di emissione diretta della fattura. Tuttavia, va ricordato che le fattispecie sanzionatorie, nel nostro ordinamento, sono tipiche: vanno quindi espressamente previste dalla legge e per esse vige il divieto di estensione analogica. Mancando una norma specifica che impedisca di fatturare direttamente al paziente, questo modo di operare deve pertanto considerarsi lecito. In linea con quanto previsto dalla circolare 39/E del 2010, deve però ritenersi che, ai fini della deducibilità della spesa da parte del cliente, la fattura vada accompagnata dalla prescrizione dell odontoiatra, presupposto indispensabile per la prestazione resa dall odontotecnico.
51
Daniele Dondarini Segreteria ANTOI
 Daniele Dondarini Segreteria ANTOI 2017/745 5 APRILE 2017 Pubblicato sulla Gazzetta Ufficiale dell Unione Europea (L117/1) del 5 maggio 2017 REGOLAMENTO DISPOSITIVI MEDICI (UE) 2017/745 (MDR) 25 MAGGIO
Daniele Dondarini Segreteria ANTOI 2017/745 5 APRILE 2017 Pubblicato sulla Gazzetta Ufficiale dell Unione Europea (L117/1) del 5 maggio 2017 REGOLAMENTO DISPOSITIVI MEDICI (UE) 2017/745 (MDR) 25 MAGGIO
Dispositivo-vigilanza: aspetti normativi e procedura aziendale per la segnalazione di incidenti o mancati incidenti da dispositivo
 Dispositivo-vigilanza: aspetti normativi e procedura aziendale per la segnalazione di incidenti o mancati incidenti da dispositivo medico Dr.ssa Liliana Ciannarella Dirigente Sanitario Farmacista A.O.Ospedale
Dispositivo-vigilanza: aspetti normativi e procedura aziendale per la segnalazione di incidenti o mancati incidenti da dispositivo medico Dr.ssa Liliana Ciannarella Dirigente Sanitario Farmacista A.O.Ospedale
PROTESI DENTARIE RICAVATE CON TECNICHE CAD-CAM
 PROTESI DENTARIE RICAVATE CON TECNICHE CAD-CAM Problematica Qualificazione giuridica del prodotto Garantire maggiore trasparenza e tracciabilità nei processi di fabbricazione dei dispositivi medici dentari
PROTESI DENTARIE RICAVATE CON TECNICHE CAD-CAM Problematica Qualificazione giuridica del prodotto Garantire maggiore trasparenza e tracciabilità nei processi di fabbricazione dei dispositivi medici dentari
LE PRESCRIVO UNA APP DAL TACCUINO AL PERSONAL HEALTH RECORD
 LE PRESCRIVO UNA APP DAL TACCUINO AL PERSONAL HEALTH RECORD m-health e Medical APPs 10 domande sulla sicurezza del software medico Alla ricerca di risposte nel futuro Regolamento Europeo sui Dispositivi
LE PRESCRIVO UNA APP DAL TACCUINO AL PERSONAL HEALTH RECORD m-health e Medical APPs 10 domande sulla sicurezza del software medico Alla ricerca di risposte nel futuro Regolamento Europeo sui Dispositivi
I GIORNATA CONGRESSUALE 17 aprile 2018
 I GIORNATA CONGRESSUALE 17 aprile 2018 Le tecniche ortopediche nel mare delle Norme, dei Regolamenti, del Mercato e dell Innovazione: le sfide per il comparto Daniele Dondarini CIDOS Emilia Romagna 2017/745
I GIORNATA CONGRESSUALE 17 aprile 2018 Le tecniche ortopediche nel mare delle Norme, dei Regolamenti, del Mercato e dell Innovazione: le sfide per il comparto Daniele Dondarini CIDOS Emilia Romagna 2017/745
Tracciabilità dei Dispositivi Medici
 Pavia, 22 febbraio 2010 Tracciabilità dei Dispositivi Medici S.C. Farmacia FONDAZIONE I.R.C.C.S. Policlinico S. Matteo Pavia Normative correnti italiane ed europee Le tre normative che regolamentano, rispettivamente,
Pavia, 22 febbraio 2010 Tracciabilità dei Dispositivi Medici S.C. Farmacia FONDAZIONE I.R.C.C.S. Policlinico S. Matteo Pavia Normative correnti italiane ed europee Le tre normative che regolamentano, rispettivamente,
SCHEDA SINOTTICA DI CONSULTAZIONE DELLA NORMATIVA
 SCHEDA SINOTTICA DI CONSULTAZIONE DELLA NORMATIVA Normativa D. Legislativo n. 507 del 14/12/1992 Titolo Attuazione della direttiva 90/385/CEE concernente il ravvicinamento delle legislazioni degli Stati
SCHEDA SINOTTICA DI CONSULTAZIONE DELLA NORMATIVA Normativa D. Legislativo n. 507 del 14/12/1992 Titolo Attuazione della direttiva 90/385/CEE concernente il ravvicinamento delle legislazioni degli Stati
GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI
 GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI Sorveglianza e Vigilanza Roberto Pozzi- AIRMEDD BD Italia SpA EXPODENTAL MEETIING 17 Maggio 2018 Vigilanza Segnalazione incidenti
GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI Sorveglianza e Vigilanza Roberto Pozzi- AIRMEDD BD Italia SpA EXPODENTAL MEETIING 17 Maggio 2018 Vigilanza Segnalazione incidenti
Legenda: D.Lgs. 46/1997. Pre D.Lgs. 37/2010 Post D.Lgs. 37/2010. Articolo 1. Articolo 1. Definizioni. Definizioni.
 Legenda: laddove una parte del testo contenuto nella colonna di destra (post D.Lgs. 37/2010) è evidenziata in giallo, essa è indicativa di una modifica ovvero integrazione del D.Lgs. 46/97 ad opera del
Legenda: laddove una parte del testo contenuto nella colonna di destra (post D.Lgs. 37/2010) è evidenziata in giallo, essa è indicativa di una modifica ovvero integrazione del D.Lgs. 46/97 ad opera del
Quale responsabilità per l infermiere e il coordinatore nel ricondizionamento dei dispositivi monouso
 Quale responsabilità per l infermiere e il coordinatore nel ricondizionamento dei dispositivi monouso Luca Benci www.lucabenci.it Twitter @Luca_Benci Rimini, 8-9 aprile 2016 L evoluzione delle leggi sulla
Quale responsabilità per l infermiere e il coordinatore nel ricondizionamento dei dispositivi monouso Luca Benci www.lucabenci.it Twitter @Luca_Benci Rimini, 8-9 aprile 2016 L evoluzione delle leggi sulla
Nuovo Regolamento europeo sui dispositivi medici: aggiornamenti sulla discussione in corso al Consiglio UE
 SIMPOSIO AFI Le imprese farmaceutiche ed i settori collegati: quale futuro? DISPOSITIVI MEDICI BORDERLINE: ASPETTI DI QUALITÀ E SICUREZZA Nuovo Regolamento europeo sui dispositivi medici: aggiornamenti
SIMPOSIO AFI Le imprese farmaceutiche ed i settori collegati: quale futuro? DISPOSITIVI MEDICI BORDERLINE: ASPETTI DI QUALITÀ E SICUREZZA Nuovo Regolamento europeo sui dispositivi medici: aggiornamenti
III CONGRESSO CONGIUNTO DI ORTOPEDIA TECNICA
 Il razionale del nuovo Regolamento III CONGRESSO CONGIUNTO DI ORTOPEDIA TECNICA Gian Luca Salerio, Dir. Area Normazione Internazionale UNI Sandro Storelli, Coordinatore Innovazione e ricerca CNA Padova
Il razionale del nuovo Regolamento III CONGRESSO CONGIUNTO DI ORTOPEDIA TECNICA Gian Luca Salerio, Dir. Area Normazione Internazionale UNI Sandro Storelli, Coordinatore Innovazione e ricerca CNA Padova
Vigilanza sui dispositivi medici
 Livorno 29 Aprile 2006 IL SISTEMA DI FARMACOVIGILANZA E IL PERCORSO DI SEGNALAZIONE DELL ADRs Vigilanza sui dispositivi medici DISPOSITIVI MEDICI Difetti di fabbricazione, Problemi occasionali, Errori
Livorno 29 Aprile 2006 IL SISTEMA DI FARMACOVIGILANZA E IL PERCORSO DI SEGNALAZIONE DELL ADRs Vigilanza sui dispositivi medici DISPOSITIVI MEDICI Difetti di fabbricazione, Problemi occasionali, Errori
PROCEDURA OPERATIVA PER LA SORVEGLIANZA
 26/01/2011 Pag. 1 di 6 PROCEDURA OPERATIVA PER LA SORVEGLIANZA POST- VENDITA 1. SCOPO... 2 2. APPLICABILITÀ... 2 3. DOCUMENTI DI RIFERIMENTO... 2 3.1. Modulistica di riferimento... 3 4. RESPONSABILITÀ...
26/01/2011 Pag. 1 di 6 PROCEDURA OPERATIVA PER LA SORVEGLIANZA POST- VENDITA 1. SCOPO... 2 2. APPLICABILITÀ... 2 3. DOCUMENTI DI RIFERIMENTO... 2 3.1. Modulistica di riferimento... 3 4. RESPONSABILITÀ...
Il ruolo dell Ente Notificato nella Marcatura CE degli Impianti
 Il ruolo dell Ente Notificato nella Marcatura CE degli Impianti Convegno gas medicinali e gli impianti di distribuzione Ancona 28 giugno 2007 Giovanni Ceriani IMPIANTO di DISTRIBUZIONE Definiamo impianto
Il ruolo dell Ente Notificato nella Marcatura CE degli Impianti Convegno gas medicinali e gli impianti di distribuzione Ancona 28 giugno 2007 Giovanni Ceriani IMPIANTO di DISTRIBUZIONE Definiamo impianto
Dispositivi medici e nuovo Regolamento Europeo
 Società di Scienze Farmacologiche Applicate Society for Applied Pharmacological Sciences Seminario GdL Dispositivi Medici 15 Luglio 2013 Milano Dispositivi medici e nuovo Regolamento Europeo Principali
Società di Scienze Farmacologiche Applicate Society for Applied Pharmacological Sciences Seminario GdL Dispositivi Medici 15 Luglio 2013 Milano Dispositivi medici e nuovo Regolamento Europeo Principali
Realizzazione. MDR Regolamento dispositivi medici Reg. (UE) 2017/745
 2017/745") Realizzazione Certifico S.r.l. Sede op.: A. De Curtis 28-06135 PERUGIA Sede amm.: Via Benedetto Croce 15-06024 Gubbio PERUGIA Tel. + 39 075 5997363 + 39 075 5997343 Assistenza 800 14 47 46 info@certifico.com
Realizzazione Certifico S.r.l. Sede op.: A. De Curtis 28-06135 PERUGIA Sede amm.: Via Benedetto Croce 15-06024 Gubbio PERUGIA Tel. + 39 075 5997363 + 39 075 5997343 Assistenza 800 14 47 46 info@certifico.com
La salute e la sicurezza del lavoro nell uso dei dispositivi medici
 Dipartimento Tecnologie di Sicurezza CONVEGNO La sicurezza degli impianti elettrici e dei dispositivi medici nelle strutture sanitarie La salute e la sicurezza del lavoro nell uso dei dispositivi medici
Dipartimento Tecnologie di Sicurezza CONVEGNO La sicurezza degli impianti elettrici e dei dispositivi medici nelle strutture sanitarie La salute e la sicurezza del lavoro nell uso dei dispositivi medici
Certifico S.r.l. IT 05/05/ di 260 MDR Regolamento dispositivi medici Reg. (UE) 2017/745 Ed
 2017/745 Ed") 05/05/2019 2 di 260 MDR Regolamento dispositivi medici Reg. (UE) 2017/745 Ed. 1.1 2019 Realizzazione Certifico S.r.l. Sede op.: A. De Curtis 28-06135 PERUGIA Sede amm.: Via Benedetto Croce 15-06024 Gubbio
05/05/2019 2 di 260 MDR Regolamento dispositivi medici Reg. (UE) 2017/745 Ed. 1.1 2019 Realizzazione Certifico S.r.l. Sede op.: A. De Curtis 28-06135 PERUGIA Sede amm.: Via Benedetto Croce 15-06024 Gubbio
Dispositivi medici: attività di sorveglianza in frontiera
 Polizia Sanitaria Roma, 20 Aprile 2011 Dispositivi medici: attività di sorveglianza in frontiera 1 dr. Franco Abbenda Ministero della Salute DGFDM Una precisazione Farmaco, cosmetico o dispositivo medico??
Polizia Sanitaria Roma, 20 Aprile 2011 Dispositivi medici: attività di sorveglianza in frontiera 1 dr. Franco Abbenda Ministero della Salute DGFDM Una precisazione Farmaco, cosmetico o dispositivo medico??
GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI
 GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI Linda Sanin UNIDI (Unione Nazionale Industrie Dentarie Italiane) EXPODENTAL MEETIING 17 Maggio 2018 DA DOVE NASCE L ESIGENZA
GLI OPERATORI ECONOMICI NEL QUADRO DEI NUOVI REGOLAMENTI SUI DISPOSITIVI MEDICI Linda Sanin UNIDI (Unione Nazionale Industrie Dentarie Italiane) EXPODENTAL MEETIING 17 Maggio 2018 DA DOVE NASCE L ESIGENZA
Seminario FederSalus. Integratori alimentari e dispositivi medici. Dinamiche a confronto: aspetti regolatori e di marketing. Luciana Gramiccioni
 Seminario FederSalus 30 settembre 2014 Milano Integratori alimentari e dispositivi medici. Dinamiche a confronto: aspetti regolatori e di marketing Luciana Gramiccioni DIRETTIVA 2007/47/CE Definizione
Seminario FederSalus 30 settembre 2014 Milano Integratori alimentari e dispositivi medici. Dinamiche a confronto: aspetti regolatori e di marketing Luciana Gramiccioni DIRETTIVA 2007/47/CE Definizione
I dispositivi medici: aspetti generali e classificatori
 Capitolo primo I dispositivi medici: aspetti generali e classificatori 1.1 Dispositivi medici: generalità e definizioni I dispositivi medici rappresentano un settore di grande interesse, se considerato
Capitolo primo I dispositivi medici: aspetti generali e classificatori 1.1 Dispositivi medici: generalità e definizioni I dispositivi medici rappresentano un settore di grande interesse, se considerato
REGOLAMENTO (UE) N. 2017/745 (MDR) GUIDA ALL APPLICAZIONE
 N. 2017/745 (MDR) GUIDA ALL APPLICAZIONE") DISPOSITIVI MEDICI (MDR) GUIDA ALL APPLICAZIONE Febbraio 2019 IMQ www.imq.it info@imq.it Indice 1 ENTRATA IN VIGORE E APPLICAZIONE pag. 5 2 DISPOSIZIONI TRANSITORIE 3 AMBITO DI APPLICAZIONE MDR pag. 6
DISPOSITIVI MEDICI (MDR) GUIDA ALL APPLICAZIONE Febbraio 2019 IMQ www.imq.it info@imq.it Indice 1 ENTRATA IN VIGORE E APPLICAZIONE pag. 5 2 DISPOSIZIONI TRANSITORIE 3 AMBITO DI APPLICAZIONE MDR pag. 6
ALLEGATO XI VALUTAZIONE DELLA CONFORMITÀ BASATA SULLA VERIFICA DELLA CONFORMITÀ DEL PRODOTTO
 ALLEGATO XI VALUTAZIONE DELLA CONFORMITÀ BASATA SULLA VERIFICA DELLA CONFORMITÀ DEL PRODOTTO 1. L'obiettivo della valutazione della conformità basata sulla verifica della conformità del prodotto è garantire
ALLEGATO XI VALUTAZIONE DELLA CONFORMITÀ BASATA SULLA VERIFICA DELLA CONFORMITÀ DEL PRODOTTO 1. L'obiettivo della valutazione della conformità basata sulla verifica della conformità del prodotto è garantire
Cosa cambia dal punto di vista dell industria
 Cosa cambia dal punto di vista dell industria Il nuovo regolamento Dispositivi Medici XVIII Convegno Nazionale AIIC -Genova, 8 Aprile 2017 1 Il testo finale dei regolamenti MD & IVD è stato votato! Il
Cosa cambia dal punto di vista dell industria Il nuovo regolamento Dispositivi Medici XVIII Convegno Nazionale AIIC -Genova, 8 Aprile 2017 1 Il testo finale dei regolamenti MD & IVD è stato votato! Il
EUROPA: ANCORA SORPRESE? Daniele Dondarini Resp. CNA Unione Benessere e Sanità Emilia-Romagna
 EUROPA: ANCORA SORPRESE? Daniele Dondarini Resp. CNA Unione Benessere e Sanità Emilia-Romagna Proposta di REGOLAMENTO DEL PARLAMENTO EUROPEO E DEL CONSIGLIO In un mercato interno cui partecipano 32 paesi
EUROPA: ANCORA SORPRESE? Daniele Dondarini Resp. CNA Unione Benessere e Sanità Emilia-Romagna Proposta di REGOLAMENTO DEL PARLAMENTO EUROPEO E DEL CONSIGLIO In un mercato interno cui partecipano 32 paesi
Situazione piccole strutture in Ticino
 Situazione piccole strutture in Ticino Anna De Benedetti, Caposervizio Vigilanza e qualità Ufficio del Medico cantonale Orlando Petrini, Direttore Istituto di microbiologia cantonale ODMed 2011 1 L Ordinanza
Situazione piccole strutture in Ticino Anna De Benedetti, Caposervizio Vigilanza e qualità Ufficio del Medico cantonale Orlando Petrini, Direttore Istituto di microbiologia cantonale ODMed 2011 1 L Ordinanza
QUESITI. 1) obbligo di consegnare al paziente la documentazione o se ci si può limitare a tenerla in studio a disposizione dello stesso; [ ]
![QUESITI. 1) obbligo di consegnare al paziente la documentazione o se ci si può limitare a tenerla in studio a disposizione dello stesso; [ ]](/thumbs/88/116535558.jpg "QUESITI. 1) obbligo di consegnare al paziente la documentazione o se ci si può limitare a tenerla in studio a disposizione dello stesso; [ ]") QUESITI a seguito del D. Lgs. 37 del 25 Gennaio scorso è entrata in vigore la Direttiva Europea 47/2007 che sostituisce, con modifiche ed integrazioni, la precedente Direttiva 93/42 [ ]. A tal proposito,
QUESITI a seguito del D. Lgs. 37 del 25 Gennaio scorso è entrata in vigore la Direttiva Europea 47/2007 che sostituisce, con modifiche ed integrazioni, la precedente Direttiva 93/42 [ ]. A tal proposito,
LINEA GUIDA ESTENSIONE DEL PERIODO DI UTILIZZO DEGLI IMPIANTI DI DISTRIBUZIONE DEI GAS MEDICINALI, DEL VUOTO E DI EVACUAZIONE DEI GAS ANESTETICI
 LINEA GUIDA ESTENSIONE DEL PERIODO DI UTILIZZO DEGLI IMPIANTI DI DISTRIBUZIONE DEI GAS MEDICINALI, DEL VUOTO E DI EVACUAZIONE DEI GAS ANESTETICI I EDIZIONE - gennaio 2019 Il presente documento è stato
LINEA GUIDA ESTENSIONE DEL PERIODO DI UTILIZZO DEGLI IMPIANTI DI DISTRIBUZIONE DEI GAS MEDICINALI, DEL VUOTO E DI EVACUAZIONE DEI GAS ANESTETICI I EDIZIONE - gennaio 2019 Il presente documento è stato
C) DATI RELATIVI ALL EVENTO
 DATI RELATIVI ALL EVENTO") Fornitore (nome, ragione sociale e indirizzo) Nome commerciale ed eventuale modello del dispositivo Descrizione del dispositivo medico N. codice del dispositivo assegnato dal fabbricante Numero di lotto
Fornitore (nome, ragione sociale e indirizzo) Nome commerciale ed eventuale modello del dispositivo Descrizione del dispositivo medico N. codice del dispositivo assegnato dal fabbricante Numero di lotto
GLI ATTORI DEL NUOVO REGOLAMENTO E LE LORO
 GLI ATTORI DEL NUOVO REGOLAMENTO E LE LORO RESPONSABILITA Alessandro Berti Vice Presidente AIRMEDD Persona responsabile del rispetto della normativa Supply chain(operatori economici) Fabbricante e Mandatario
GLI ATTORI DEL NUOVO REGOLAMENTO E LE LORO RESPONSABILITA Alessandro Berti Vice Presidente AIRMEDD Persona responsabile del rispetto della normativa Supply chain(operatori economici) Fabbricante e Mandatario
PERSONA RESPONSABILE UNA NUOVA FIGURA PROFESSIONALE IN AZIENDA. Dott.ssa Cristina Bazzaro QA & RA Manager
 PERSONA RESPONSABILE UNA NUOVA FIGURA PROFESSIONALE IN AZIENDA Dott.ssa Cristina Bazzaro QA & RA Manager Articolo 15 Persona responsabile del rispetto della normativa 1.I fabbricanti, all'interno della
PERSONA RESPONSABILE UNA NUOVA FIGURA PROFESSIONALE IN AZIENDA Dott.ssa Cristina Bazzaro QA & RA Manager Articolo 15 Persona responsabile del rispetto della normativa 1.I fabbricanti, all'interno della
DECRETO LEGISLATIVO 9 APRILE 2008, N. 81
 DECRETO LEGISLATIVO 9 APRILE 2008, N. 81 Testo coordinato con il Decreto Legislativo 3 agosto 2009, n. 106 Attuazione dell'articolo 1 della legge 3 agosto 2007, n. 123, in materia di tutela della salute
DECRETO LEGISLATIVO 9 APRILE 2008, N. 81 Testo coordinato con il Decreto Legislativo 3 agosto 2009, n. 106 Attuazione dell'articolo 1 della legge 3 agosto 2007, n. 123, in materia di tutela della salute
Dott. Roberto Baldini Dirigente Farmacista Responsabile S.S. Gestione Dispositivi Medici U. O. C. Farmacia IRCCS A.O.U. San Martino IST Genova
 Corso aggiornamento sezione regionale SIFO Liguria Dott. Roberto Baldini Dirigente Farmacista Responsabile S.S. Gestione Dispositivi Medici U. O. C. Farmacia IRCCS A.O.U. San Martino IST Genova Corso SIFO
Corso aggiornamento sezione regionale SIFO Liguria Dott. Roberto Baldini Dirigente Farmacista Responsabile S.S. Gestione Dispositivi Medici U. O. C. Farmacia IRCCS A.O.U. San Martino IST Genova Corso SIFO
E LA SICUREZZA DELLE APPARECCHIATURE ELETTROMEDICALI E DEGLI IMPIANTI
 Il D.Lgs. 81/2008 il D.Lgs. 106/2009, le Norme Europee E LA SICUREZZA DELLE APPARECCHIATURE ELETTROMEDICALI E DEGLI IMPIANTI Vincenzo Ventimigla IL D.LGS. 81/2008, il D.Lgs. 106/2009 e le attrezzature
Il D.Lgs. 81/2008 il D.Lgs. 106/2009, le Norme Europee E LA SICUREZZA DELLE APPARECCHIATURE ELETTROMEDICALI E DEGLI IMPIANTI Vincenzo Ventimigla IL D.LGS. 81/2008, il D.Lgs. 106/2009 e le attrezzature
DISPOSITIVI MEDICI E LA GESTIONE DEL RISCHIO
 ANTE riccione 17 aprile 2012 DISPOSITIVI MEDICI E LA GESTIONE DEL RISCHIO RUOLO E INTERAZIONE DEI SOGGETTI OPERANTI NELLA DIRETTIVA E RELATIVE RESPONSABILITA avv. Silvia Stefanelli 1 DIR 93/42/CEE Direttiva
ANTE riccione 17 aprile 2012 DISPOSITIVI MEDICI E LA GESTIONE DEL RISCHIO RUOLO E INTERAZIONE DEI SOGGETTI OPERANTI NELLA DIRETTIVA E RELATIVE RESPONSABILITA avv. Silvia Stefanelli 1 DIR 93/42/CEE Direttiva
I PIANI DI CONTROLLO DEGLI APPARECCHI DI SOLLEVAMENTO MATERIALI MILANO 3 LUGLIO 2014 ATTREZZATURE DI SOLLEVAMENTO: RUOLI E RESPONSABILITÀ
 I PIANI DI CONTROLLO DEGLI APPARECCHI DI SOLLEVAMENTO MATERIALI MILANO 3 LUGLIO 2014 ATTREZZATURE DI SOLLEVAMENTO: RUOLI E RESPONSABILITÀ SCHEDE PER LA DEFINIZIONE DI PIANI PER I CONTROLLI DI APPARECCHI
I PIANI DI CONTROLLO DEGLI APPARECCHI DI SOLLEVAMENTO MATERIALI MILANO 3 LUGLIO 2014 ATTREZZATURE DI SOLLEVAMENTO: RUOLI E RESPONSABILITÀ SCHEDE PER LA DEFINIZIONE DI PIANI PER I CONTROLLI DI APPARECCHI
La normativa europea dei dispositivi medici e il ruolo del Regulatory Affairs Manager
 La normativa europea dei dispositivi medici e il ruolo del Regulatory Affairs Manager Antonella Mamoli AFI Università degli Studi di Torino 16 Maggio 2014 AGENDA: 1. Chi è il Regulatory Affairs Manager
La normativa europea dei dispositivi medici e il ruolo del Regulatory Affairs Manager Antonella Mamoli AFI Università degli Studi di Torino 16 Maggio 2014 AGENDA: 1. Chi è il Regulatory Affairs Manager
Franco Gerardi Pavia - 17 maggio 2013
 Franco Gerardi Pavia - 17 maggio 2013 Sicurezza Normativa e responsabilità Ogni attività lavorativa deve rispettare alcune norme italiane (derivate da direttive europee) che definiscono le attività di
Franco Gerardi Pavia - 17 maggio 2013 Sicurezza Normativa e responsabilità Ogni attività lavorativa deve rispettare alcune norme italiane (derivate da direttive europee) che definiscono le attività di
ESEMPIO DI CONTRATTO DI DISTRIBUZIONE / IMPORTAZIONE 1
 ESEMPIO DI CONTRATTO DI DISTRIBUZIONE / IMPORTAZIONE 1 (di seguito ), una filiale interamente di proprietà di., entrambe le società interamente di proprietà di ASSICURAZIONE DEL CONTROLLO QUALITA, VIGILANZA
ESEMPIO DI CONTRATTO DI DISTRIBUZIONE / IMPORTAZIONE 1 (di seguito ), una filiale interamente di proprietà di., entrambe le società interamente di proprietà di ASSICURAZIONE DEL CONTROLLO QUALITA, VIGILANZA
PROTOCOLLO INDAGINI CLINICHE CON DISPOSITIVO MEDICO
 Mod. B10 Vers_20160118 PROTOCOLLO INDAGINI CLINICHE CON DISPOSITIVO MEDICO Titolo dello Studio: (Dovrebbe descrivere il disegno dello studio, la popolazione, gli interventi, e se applicabile, riporta l
Mod. B10 Vers_20160118 PROTOCOLLO INDAGINI CLINICHE CON DISPOSITIVO MEDICO Titolo dello Studio: (Dovrebbe descrivere il disegno dello studio, la popolazione, gli interventi, e se applicabile, riporta l
Sommario PARTE I - T.U. N. 81/2008
 XI Presentazione... Profili Autori... V VII PARTE I - T.U. N. 81/2008 CAPITOLO 1 TITOLO I T.U. - PRINCIPI COMUNI CAPO I - DISPOSIZIONI GENERALI... 1 Il lavoratore... 6 Il datore di lavoro... 8 Il dirigente...
XI Presentazione... Profili Autori... V VII PARTE I - T.U. N. 81/2008 CAPITOLO 1 TITOLO I T.U. - PRINCIPI COMUNI CAPO I - DISPOSIZIONI GENERALI... 1 Il lavoratore... 6 Il datore di lavoro... 8 Il dirigente...
Il datore di lavoro: obblighi di controllo, verifica e formazione
 INAIL ASSOLOMBARDA Sorveglianza del mercato, verifica periodica e formazione sugli apparecchi di sollevamento: sinergie per la sicurezza Il datore di lavoro: obblighi di controllo, verifica e formazione
INAIL ASSOLOMBARDA Sorveglianza del mercato, verifica periodica e formazione sugli apparecchi di sollevamento: sinergie per la sicurezza Il datore di lavoro: obblighi di controllo, verifica e formazione
Ministero della Salute
 Ministero della Salute DECRETO 15 novembre 2005 Approvazione dei modelli di schede di segnalazioni di incidenti o mancati incidenti, che coinvolgono dispositivi medici e dispositivi medico-diagnostici
Ministero della Salute DECRETO 15 novembre 2005 Approvazione dei modelli di schede di segnalazioni di incidenti o mancati incidenti, che coinvolgono dispositivi medici e dispositivi medico-diagnostici
NUOVO REGOLAMENTO UE SUI DISPOSITIVI MEDICI
 NUOVO REGOLAMENTO UE SUI DISPOSITIVI MEDICI Come prepararsi alla deadline 2020 Ramada Plaza Milano, Via Stamira d Ancona 27, Milano VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE NEL NUOVO REGOLAMENTO
NUOVO REGOLAMENTO UE SUI DISPOSITIVI MEDICI Come prepararsi alla deadline 2020 Ramada Plaza Milano, Via Stamira d Ancona 27, Milano VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE NEL NUOVO REGOLAMENTO
Caserta 12 Giugno 2006 RIFERIMENTI NORMATIVI. Relatore ing. Giuseppe ESPOSITO
 Caserta 12 Giugno 2006 RIFERIMENTI NORMATIVI Relatore ing. Giuseppe ESPOSITO LE PROBLEMATICHE AUTORIZZATIVE CONCERNENTI LA PREVENZIONE E LA SICUREZZA DEI LUOGHI DI LAVORO Prim ancora di procedere con la
Caserta 12 Giugno 2006 RIFERIMENTI NORMATIVI Relatore ing. Giuseppe ESPOSITO LE PROBLEMATICHE AUTORIZZATIVE CONCERNENTI LA PREVENZIONE E LA SICUREZZA DEI LUOGHI DI LAVORO Prim ancora di procedere con la
LA PERSONA RESPONSABILE DEL RISPETTO DELLA NORMATIVA
 LA PERSONA RESPONSABILE DEL RISPETTO DELLA NORMATIVA Alessandro Berti Vice Presidente AIRMEDD Article 15 Person responsible for regulatory compliance I fabbricanti, (.mandatari ) all'interno della loro
LA PERSONA RESPONSABILE DEL RISPETTO DELLA NORMATIVA Alessandro Berti Vice Presidente AIRMEDD Article 15 Person responsible for regulatory compliance I fabbricanti, (.mandatari ) all'interno della loro
SCHEMA per la certificazione dei tubi di rame senza saldatura per condizionamento e refrigerazione cui alla norma UNI EN :2010
 SCHEMA per la certificazione dei tubi di rame senza saldatura per condizionamento e refrigerazione cui alla norma UNI EN 12735-1:2010 Il presente documento è stato approvato dalla Commissione Prodotti
SCHEMA per la certificazione dei tubi di rame senza saldatura per condizionamento e refrigerazione cui alla norma UNI EN 12735-1:2010 Il presente documento è stato approvato dalla Commissione Prodotti
Gazzetta ufficiale dell'unione europea
 L 24/6 IT 30.1.2016 REGOLAMENTO DI ESECUZIONE (UE) 2016/125 DELLA COMMISSIONE del 29 gennaio 2016 che approva il PHMB (1600; 1.8) come principio attivo esistente destinato a essere utilizzato nei biocidi
L 24/6 IT 30.1.2016 REGOLAMENTO DI ESECUZIONE (UE) 2016/125 DELLA COMMISSIONE del 29 gennaio 2016 che approva il PHMB (1600; 1.8) come principio attivo esistente destinato a essere utilizzato nei biocidi
TITOLO IX (ART ) TITOLO VII (ART )
 TITOLO VII (ART )") TITOLO VII (ART. 172-179) ATTREZZATURE MUNITE DI VIDEOTERMINALI TITOLO VIII (ART. 180-220) AGENTI FISICI PROTEZIONE DEI LAVORATORI CONTRO I RISCHI DI ESPOSIZIONE AL RUMORE DURANTE IL LAVORO PROTEZIONE
TITOLO VII (ART. 172-179) ATTREZZATURE MUNITE DI VIDEOTERMINALI TITOLO VIII (ART. 180-220) AGENTI FISICI PROTEZIONE DEI LAVORATORI CONTRO I RISCHI DI ESPOSIZIONE AL RUMORE DURANTE IL LAVORO PROTEZIONE
Rischi connessi all uso di Macchine e Attrezzature di lavoro
 Rischi connessi all uso di Macchine e Attrezzature di lavoro D. Lgs. 81-2008 Titolo III CAPO I -Articolo 69 - Definizioni 1. Agli effetti delle disposizioni di cui al presente Titolo si intende per: a)
Rischi connessi all uso di Macchine e Attrezzature di lavoro D. Lgs. 81-2008 Titolo III CAPO I -Articolo 69 - Definizioni 1. Agli effetti delle disposizioni di cui al presente Titolo si intende per: a)
Il Ministro delle Infrastrutture e dei Trasporti
 Il Ministro delle Infrastrutture e dei Trasporti VISTO il Regolamento n. 44 della Commissione economica per l Europa delle Nazioni Unite (UN/ECE) del 10 novembre 2010 recante: Disposizioni uniformi relative
Il Ministro delle Infrastrutture e dei Trasporti VISTO il Regolamento n. 44 della Commissione economica per l Europa delle Nazioni Unite (UN/ECE) del 10 novembre 2010 recante: Disposizioni uniformi relative
Progetto Regionale REACH Linea assistenza
 Progetto Regionale REACH Linea assistenza Treviso, 25 giugno 2013 1 In Italia la salute e la sicurezza sul lavoro sono regolamentate dal Decreto Legislativo n. 81 del 9 aprile 2008, anche noto come Testo
Progetto Regionale REACH Linea assistenza Treviso, 25 giugno 2013 1 In Italia la salute e la sicurezza sul lavoro sono regolamentate dal Decreto Legislativo n. 81 del 9 aprile 2008, anche noto come Testo
PRESCRIZIONI PARTICOLARI PER LA DIRETTIVA 2014/34/UE
 Titolo PRESCRIZIONI PARTICOLARI PER LA DIRETTIVA 2014/34/UE Apparecchi e sistemi di protezione per atmosfera potenzialmente esplosiva Allegato VI - MODULO C1 Conformità al Tipo basata sul controllo interno
Titolo PRESCRIZIONI PARTICOLARI PER LA DIRETTIVA 2014/34/UE Apparecchi e sistemi di protezione per atmosfera potenzialmente esplosiva Allegato VI - MODULO C1 Conformità al Tipo basata sul controllo interno
Camera di Commercio di Verona 14 aprile Relatore Gabriele Lualdi
 Camera di Commercio di Verona 14 aprile 2011 Relatore Gabriele Lualdi 128 documento, stabilito mediante consenso e approvato da un organismo riconosciuto, che fornisce, per utilizzi comuni e ripetuti,
Camera di Commercio di Verona 14 aprile 2011 Relatore Gabriele Lualdi 128 documento, stabilito mediante consenso e approvato da un organismo riconosciuto, che fornisce, per utilizzi comuni e ripetuti,
Rapporto interno n.. Rapporto relativo a:
 ALLEGATO n. 3 Rapporto di incidente o di mancato incidente da parte di operatori sanitari (artt. 9 e 10, D.Lgs. n. 46 del 1997; art. 11, D.Lgs. n. 507 del 1992) Rapporto interno n.. Rapporto relativo a:
ALLEGATO n. 3 Rapporto di incidente o di mancato incidente da parte di operatori sanitari (artt. 9 e 10, D.Lgs. n. 46 del 1997; art. 11, D.Lgs. n. 507 del 1992) Rapporto interno n.. Rapporto relativo a:
L applicazione del D.P.R. 462/01 ai locali medici
 Istituto Nazionale per l Assicurazione contro gli Infortuni sul Lavoro L applicazione del D.P.R. 462/01 ai locali medici Ing. Maria Teresa Settino Dipartimento Innovazioni Tecnologiche e Sicurezza degli
Istituto Nazionale per l Assicurazione contro gli Infortuni sul Lavoro L applicazione del D.P.R. 462/01 ai locali medici Ing. Maria Teresa Settino Dipartimento Innovazioni Tecnologiche e Sicurezza degli
Il monitoraggio continuo dei dispositivi medici
 Dipartimento Tecnologie di Sicurezza La sicurezza degli impianti elettrici e dei dispositivi medici nelle strutture sanitarie Il monitoraggio continuo dei dispositivi medici Osservatorio Salute lavoro
Dipartimento Tecnologie di Sicurezza La sicurezza degli impianti elettrici e dei dispositivi medici nelle strutture sanitarie Il monitoraggio continuo dei dispositivi medici Osservatorio Salute lavoro
PROGRAMMA CORSO PER ASPP MODULO A
 PROGRAMMA CORSO PER ASPP MODULO A Titolo Argomenti Durata Presentazione del corso L approccio alla prevenzione attraverso il D. Lgs. 626/94 per un percorso di miglioramento della sicurezza e della salute
PROGRAMMA CORSO PER ASPP MODULO A Titolo Argomenti Durata Presentazione del corso L approccio alla prevenzione attraverso il D. Lgs. 626/94 per un percorso di miglioramento della sicurezza e della salute
RISOLUZIONE N. 87/E QUESITO
 RISOLUZIONE N. 87/E Direzione Centrale Normativa e Contenzioso Roma, 12 luglio 2006 OGGETTO: istanza di Interpello - Art. 10, n. 18), del decreto del Presidente della Repubblica 29 ottobre 1972, n. 633.
RISOLUZIONE N. 87/E Direzione Centrale Normativa e Contenzioso Roma, 12 luglio 2006 OGGETTO: istanza di Interpello - Art. 10, n. 18), del decreto del Presidente della Repubblica 29 ottobre 1972, n. 633.
Il Ministro della Salute
 Il Ministro della Salute Nuove modalità per gli adempimenti previsti dall articolo 13 del decreto legislativo 24 febbraio 1997, n. 46 e successive modificazioni e per la registrazione dei dispositivi impiantabili
Il Ministro della Salute Nuove modalità per gli adempimenti previsti dall articolo 13 del decreto legislativo 24 febbraio 1997, n. 46 e successive modificazioni e per la registrazione dei dispositivi impiantabili
Seminario Tecnico Formativo
 Seminario Tecnico Formativo Classificazione Nazionale dei Dispositivi - metodologia di modifiche ed aggiornamenti Documento d indirizzo per la stesura di capitolati di gara per l acquisizione di dispositivi
Seminario Tecnico Formativo Classificazione Nazionale dei Dispositivi - metodologia di modifiche ed aggiornamenti Documento d indirizzo per la stesura di capitolati di gara per l acquisizione di dispositivi
Commissione per l'occupazione e gli affari sociali PROGETTO DI PARERE. della commissione per l'occupazione e gli affari sociali
 PARLAMENTO EUROPEO 2009-2014 Commissione per l'occupazione e gli affari sociali 11.3.2013 2012/0267(COD) PROGETTO DI PARERE della commissione per l'occupazione e gli affari sociali destinato alla commissione
PARLAMENTO EUROPEO 2009-2014 Commissione per l'occupazione e gli affari sociali 11.3.2013 2012/0267(COD) PROGETTO DI PARERE della commissione per l'occupazione e gli affari sociali destinato alla commissione
ALLEGATO 1 STUDI MEDICI ED ODONTOIATRICI: MODALITA DI PRESENTAZIONE DELLA DOMANDA DI AUTORIZZAZIONE E SCIA A) AUTORIZZAZIONE
 AUTORIZZAZIONE") STUDI MEDICI ED ODONTOIATRICI: MODALITA DI PRESENTAZIONE DELLA DOMANDA DI AUTORIZZAZIONE E SCIA A) AUTORIZZAZIONE Il professionista interessato presenta la domanda di autorizzazione al comune nel territorio
STUDI MEDICI ED ODONTOIATRICI: MODALITA DI PRESENTAZIONE DELLA DOMANDA DI AUTORIZZAZIONE E SCIA A) AUTORIZZAZIONE Il professionista interessato presenta la domanda di autorizzazione al comune nel territorio
Modalità di erogazione delle prestazioni di assistenza protesica. Art. 1 Procedura di erogazione
 Modalità di erogazione delle prestazioni di assistenza protesica ALLEGATO 12 Art. 1 Procedura di erogazione 1. La procedura di erogazione dell assistenza protesica si articola nelle seguenti fasi: formulazione
Modalità di erogazione delle prestazioni di assistenza protesica ALLEGATO 12 Art. 1 Procedura di erogazione 1. La procedura di erogazione dell assistenza protesica si articola nelle seguenti fasi: formulazione
ARPAE Agenzia regionale per la prevenzione, l'ambiente e l'energia dell'emilia - Romagna * * * Atti amministrativi
 ARPAE Agenzia regionale per la prevenzione, l'ambiente e l'energia dell'emilia - Romagna * * * Atti amministrativi Determinazione dirigenziale n. DET-AMB-2017-2547 del 19/05/2017 Oggetto Atto di adozione
ARPAE Agenzia regionale per la prevenzione, l'ambiente e l'energia dell'emilia - Romagna * * * Atti amministrativi Determinazione dirigenziale n. DET-AMB-2017-2547 del 19/05/2017 Oggetto Atto di adozione
IL DECRETO CORRETTIVO DEL D.LGS N. 81/08. Titolo III Possibili futuri scenari nell uso delle macchine. Ing. Giuseppe Piegari Ministero del lavoro
 IL DECRETO CORRETTIVO DEL D.LGS N. 81/08 Titolo III Possibili futuri scenari nell uso delle macchine Ing. Giuseppe Piegari Ministero del lavoro ART. 69 - Definizioni Agli effetti delle disposizioni di
IL DECRETO CORRETTIVO DEL D.LGS N. 81/08 Titolo III Possibili futuri scenari nell uso delle macchine Ing. Giuseppe Piegari Ministero del lavoro ART. 69 - Definizioni Agli effetti delle disposizioni di
CONTESTO DM ed i IVD sul territorio sono oltre miliardi di Euro in ricerca il 6-8% delle vendite annue di DM 10% di quelle di IVD
 Sandro Storelli Coordinatore Osservatorio Biomedicale Veneto Padova, 5 giugno 2017 REGOLAMENTAZIONE EUROPEA Per immettere in commercio i DM non è richiesta autorizzazione: il fabbricante deve effettuare
Sandro Storelli Coordinatore Osservatorio Biomedicale Veneto Padova, 5 giugno 2017 REGOLAMENTAZIONE EUROPEA Per immettere in commercio i DM non è richiesta autorizzazione: il fabbricante deve effettuare
Datore di lavoro. Dirigente
 Datore di lavoro Dirigente Medico competente RSPP Incaricati soccorso RLS Incaricato antincendio Preposto Lavoratore DATORE DI LAVORO CHI E : soggetto titolare del rapporto di lavoro con il lavoratore
Datore di lavoro Dirigente Medico competente RSPP Incaricati soccorso RLS Incaricato antincendio Preposto Lavoratore DATORE DI LAVORO CHI E : soggetto titolare del rapporto di lavoro con il lavoratore
Modalità di erogazione dei dispositivi medici monouso. Art. 1 Procedura di erogazione
 ALLEGATO 11 Modalità di erogazione dei dispositivi medici monouso Art. 1 Procedura di erogazione 1. La prescrizione dei dispositivi, effettuata sul ricettario standardizzato del Servizio sanitario nazionale,
ALLEGATO 11 Modalità di erogazione dei dispositivi medici monouso Art. 1 Procedura di erogazione 1. La prescrizione dei dispositivi, effettuata sul ricettario standardizzato del Servizio sanitario nazionale,
REGOLAMENTO PER L ATTUAZIONE DELLA DIRETTIVA 90/396/CEE, CONCERNENTE GLI APPARECCHI A GAS
 Decreto del Presidente della Repubblica 15 novembre 1996, n. 661 REGOLAMENTO PER L ATTUAZIONE DELLA DIRETTIVA 90/396/CEE, CONCERNENTE GLI APPARECCHI A GAS Art. 1 - Campo di applicazione e definizioni -
Decreto del Presidente della Repubblica 15 novembre 1996, n. 661 REGOLAMENTO PER L ATTUAZIONE DELLA DIRETTIVA 90/396/CEE, CONCERNENTE GLI APPARECCHI A GAS Art. 1 - Campo di applicazione e definizioni -
Datore di lavoro. Dirigente RSPP RLS. Preposto DATORE DI LAVORO
 Datore di lavoro Dirigente Medico competente RSPP Incaricati soccorso RLS Incaricato antincendio Preposto Lavoratore DATORE DI LAVORO CHI E : soggetto titolare del rapporto di lavoro con il lavoratore
Datore di lavoro Dirigente Medico competente RSPP Incaricati soccorso RLS Incaricato antincendio Preposto Lavoratore DATORE DI LAVORO CHI E : soggetto titolare del rapporto di lavoro con il lavoratore
GUANTI DISPOSITIVI MEDICI GUANTI DISPOSITIVI DI PROTEZIONE INDIVIDUALE
 GUANTI DISPOSITIVI MEDICI GUANTI DISPOSITIVI DI PROTEZIONE INDIVIDUALE Aspetti Normativi da Direttive a Regolamenti DISPOSITIVI MEDICI Direttiva di riferimento 93/42/CEE e D.lgs. 46 del 24/02/1997 e s.m.i.
GUANTI DISPOSITIVI MEDICI GUANTI DISPOSITIVI DI PROTEZIONE INDIVIDUALE Aspetti Normativi da Direttive a Regolamenti DISPOSITIVI MEDICI Direttiva di riferimento 93/42/CEE e D.lgs. 46 del 24/02/1997 e s.m.i.
ALLEGATO A PARTE 1 INTRODUZIONE GENERALE. Le presenti linee guida hanno i seguenti obiettivi:
 ALLEGATO A Linee guida per l efficacia delle procedure di autorizzazione e ispezione ambientale e per l introduzione di semplificazioni delle procedure amministrative per le organizzazioni in possesso
ALLEGATO A Linee guida per l efficacia delle procedure di autorizzazione e ispezione ambientale e per l introduzione di semplificazioni delle procedure amministrative per le organizzazioni in possesso
IL DLgs 81/08: QUALI OBBLIGHI? leo morisi
 IL DLgs 81/08: QUALI OBBLIGHI? leo morisi 1 Struttura Il DLgs 81/08 è composto da 305 articoli, 51 allegati + di 20 decreti o linee guida o regolamenti delegati Abroga: Il DPR 547/55 Il DPR 164/56 Il DPR
IL DLgs 81/08: QUALI OBBLIGHI? leo morisi 1 Struttura Il DLgs 81/08 è composto da 305 articoli, 51 allegati + di 20 decreti o linee guida o regolamenti delegati Abroga: Il DPR 547/55 Il DPR 164/56 Il DPR
DIREZIONE REGIONALE PER LA LOMBARDIA Direttore Regionale: Dott. Antonio Traficante
 DIREZIONE REGIONALE PER LA LOMBARDIA Direttore Regionale: Dott. Antonio Traficante SETTORE RICERCA, CERTIFICAZIONE E VERIFICA Unità operativa territoriale di Milano Direttore UOT: Dott. Ing. Michele De
DIREZIONE REGIONALE PER LA LOMBARDIA Direttore Regionale: Dott. Antonio Traficante SETTORE RICERCA, CERTIFICAZIONE E VERIFICA Unità operativa territoriale di Milano Direttore UOT: Dott. Ing. Michele De
4.10 PROVE, CONTROLLI E COLLAUDI
 Unione Industriale 55 di 94 4.10 PROVE, CONTROLLI E COLLAUDI 4.10.1 Generalità Il fornitore deve predisporre e mantenere attive procedure documentate per le attività di prova, controllo e collaudo allo
Unione Industriale 55 di 94 4.10 PROVE, CONTROLLI E COLLAUDI 4.10.1 Generalità Il fornitore deve predisporre e mantenere attive procedure documentate per le attività di prova, controllo e collaudo allo
Dispositivi di protezione individuale di I categoria
 Ministero dello Sviluppo Economico Direzione Generale per il mercato, la concorrenza, il consumatore, la vigilanza e la normativa tecnica Divisione XVI Sicurezza e conformità Dispositivi di protezione
Ministero dello Sviluppo Economico Direzione Generale per il mercato, la concorrenza, il consumatore, la vigilanza e la normativa tecnica Divisione XVI Sicurezza e conformità Dispositivi di protezione
COMMISSIONE DELLE COMUNITÀ EUROPEE. Progetto. REGOLAMENTO (UE) n. /2011 DELLA COMMISSIONE
 n. /2011 DELLA COMMISSIONE") COMMISSIONE DELLE COMUNÀ EUROPEE Progetto Bruxelles, C REGOLAMENTO (UE) n. /2011 DELLA COMMISSIONE del [ ] che modifica il regolamento (CE) n. 2042/2003 della Commissione sul mantenimento della navigabilità
COMMISSIONE DELLE COMUNÀ EUROPEE Progetto Bruxelles, C REGOLAMENTO (UE) n. /2011 DELLA COMMISSIONE del [ ] che modifica il regolamento (CE) n. 2042/2003 della Commissione sul mantenimento della navigabilità
Parte Speciale 4 Protocolli in materia di prevenzione e tutela ambientale (art. 25-undecies, D. Lgs. 231/2001)
") Parte Speciale 4 Protocolli in materia di prevenzione e tutela (art. 25-undecies, D. Lgs. 231/2001) Allegato n. 7 al Modello di Organizzazione, Gestione e Controllo approvato con determina dell Amministratore
Parte Speciale 4 Protocolli in materia di prevenzione e tutela (art. 25-undecies, D. Lgs. 231/2001) Allegato n. 7 al Modello di Organizzazione, Gestione e Controllo approvato con determina dell Amministratore
RICHIESTA DI AVVIO DEL PROCEDIMENTO UNICO DEL SUAP PER:
 DOMANDA SUAP SEMPLIFICATA Bollo in valore corrente Prot. Gen. AL SERVIZIO ATTIVITA ECONOMICHE SUAP DEL COMUNE DI CASALECCHIO DI RENO Via dei Mille, 9 40033 Casalecchio di Reno (BO) telefono: 051 598 229
DOMANDA SUAP SEMPLIFICATA Bollo in valore corrente Prot. Gen. AL SERVIZIO ATTIVITA ECONOMICHE SUAP DEL COMUNE DI CASALECCHIO DI RENO Via dei Mille, 9 40033 Casalecchio di Reno (BO) telefono: 051 598 229
SEGNALAZIONE CERTIFICATA DI INIZIO ATTIVITA (S.C.I.A.)
") AL COMUNE DI MONTE SAN SAVINO U Sportello Unico per le Attività Produttive Corso Sangallo, 38-52048 Monte San Savino (Arezzo) Da inoltrare utilizzando il Portale Regionale accessibile dal sito web del
AL COMUNE DI MONTE SAN SAVINO U Sportello Unico per le Attività Produttive Corso Sangallo, 38-52048 Monte San Savino (Arezzo) Da inoltrare utilizzando il Portale Regionale accessibile dal sito web del
Ing. Francesca Ferrocci Bologna, 17 ottobre Area Sicurezza delle costruzioni - ANCE
 Ing. Francesca Ferrocci Bologna, 17 ottobre 2013 Area Sicurezza delle costruzioni - ANCE GLI INFORTUNI NEL SETTORE COSTRUZIONI (CODICE ATECO ISTAT 2007 «F») 2 3 4 5 Obblighi del datore di lavoro L obbligo
Ing. Francesca Ferrocci Bologna, 17 ottobre 2013 Area Sicurezza delle costruzioni - ANCE GLI INFORTUNI NEL SETTORE COSTRUZIONI (CODICE ATECO ISTAT 2007 «F») 2 3 4 5 Obblighi del datore di lavoro L obbligo
Secondo il Nuovo Regolamento i DPI sono di seguito definiti (art. 3 comma 1) come:
 come:") NUOVO REGOLAMENTO EUROPEO SUI DISPOSITIVI DI PROTEZIONE INDIVIDUALE (DPI) A far data dal 21 aprile 2018 si applicherà il nuovo Regolamento UE 2016/425 sui Dispositivi di Protezione Individuale che abroga
NUOVO REGOLAMENTO EUROPEO SUI DISPOSITIVI DI PROTEZIONE INDIVIDUALE (DPI) A far data dal 21 aprile 2018 si applicherà il nuovo Regolamento UE 2016/425 sui Dispositivi di Protezione Individuale che abroga
Direttiva 2006/121/CE del Parlamento europeo e del Consiglio. del 18 dicembre 2006
 L 396/850 IT Gazzetta ufficiale dell Unione europea 30.12.2006 Direttiva 2006/121/CE del Parlamento europeo e del Consiglio del 18 dicembre 2006 che modifica la direttiva 67/548/CEE del Consiglio concernente
L 396/850 IT Gazzetta ufficiale dell Unione europea 30.12.2006 Direttiva 2006/121/CE del Parlamento europeo e del Consiglio del 18 dicembre 2006 che modifica la direttiva 67/548/CEE del Consiglio concernente
ALLEGATO n. 5 RAPPORTO INIZIALE sulle alterazioni delle caratteristiche e delle prestazioni
 ALLEGATO n. 5 RAPPORTO INIZIALE sulle alterazioni delle caratteristiche e delle prestazioni di un dispositivo medico-diagnostico in vitro da parte del fabbricante al Ministero della Salute (art. 11, D.Lgs.
ALLEGATO n. 5 RAPPORTO INIZIALE sulle alterazioni delle caratteristiche e delle prestazioni di un dispositivo medico-diagnostico in vitro da parte del fabbricante al Ministero della Salute (art. 11, D.Lgs.
Rapporto relativo a: Incidente. A) Dati relativi al luogo dove si è verificato l'episodio
 Dati relativi al luogo dove si è verificato l'episodio") RAPPORTO I INCIENTE A PARTE I OPERA TORI SANITARI AL MINISTERO ELLA SALUTE (artt. 9 e 10,.Lgs. n.46 del 1997; art.11,.lgs. n. 507 del 1992, artt. 1 e 9,.Lgs. n. 37 del 2010) Rapporto relativo a: Incidente
RAPPORTO I INCIENTE A PARTE I OPERA TORI SANITARI AL MINISTERO ELLA SALUTE (artt. 9 e 10,.Lgs. n.46 del 1997; art.11,.lgs. n. 507 del 1992, artt. 1 e 9,.Lgs. n. 37 del 2010) Rapporto relativo a: Incidente
COORDINATORE ALLA SICUREZZA IN FASE DI PROGETTAZIONE E ESECUZIONE
 COORDINATORE ALLA SICUREZZA IN FASE DI PROGETTAZIONE E ESECUZIONE MACCHINE E ATTREZZATURE IN CANTIERE: GLI OBBLIGHI PER IL COORDINATORE PER LA SICUREZZA - I PARTE - GEOM. LUCA PERLATI VERONA, LÌ 02/05/2018
COORDINATORE ALLA SICUREZZA IN FASE DI PROGETTAZIONE E ESECUZIONE MACCHINE E ATTREZZATURE IN CANTIERE: GLI OBBLIGHI PER IL COORDINATORE PER LA SICUREZZA - I PARTE - GEOM. LUCA PERLATI VERONA, LÌ 02/05/2018
ISTITUTO TUMORI GIOVANNI PAOLO II ISTITUTO DI RICOVERO E CURA A CARATTERE SCIENTIFICO. Deliberazione del Direttore Generale n.
 ISTITUTO TUMORI GIOVANNI PAOLO II ISTITUTO DI RICOVERO E CURA A CARATTERE SCIENTIFICO BARI Deliberazione del Direttore Generale n. 546 del registro OGGETTO: PROCEDURA PER LA RICHIESTA DI DISPOSITIVI MEDICI
ISTITUTO TUMORI GIOVANNI PAOLO II ISTITUTO DI RICOVERO E CURA A CARATTERE SCIENTIFICO BARI Deliberazione del Direttore Generale n. 546 del registro OGGETTO: PROCEDURA PER LA RICHIESTA DI DISPOSITIVI MEDICI
I CONTROLLI DOCUMENTALI SUI GIOCATTOLI
 I CONTROLLI DOCUMENTALI SUI GIOCATTOLI A P P R O F O N D I M E N T O SUI C O N T R O L L I D O C U M E N T A L I N E L L A M B I T O D E L P I A N O F O R M A T I V O IN M A T E R I A DI V I G I L A N
I CONTROLLI DOCUMENTALI SUI GIOCATTOLI A P P R O F O N D I M E N T O SUI C O N T R O L L I D O C U M E N T A L I N E L L A M B I T O D E L P I A N O F O R M A T I V O IN M A T E R I A DI V I G I L A N
Impianti elettrico. Evidenza SI NO NA Riferimenti Note
 Impianti elettrico Per tutti i locali dell'unità Operativa in considerazione, è disponibile la classificazione aggiornata, firmata dal Responsabile Sanitario Norma CEI (ambiente ordinario, locale ad uso
Impianti elettrico Per tutti i locali dell'unità Operativa in considerazione, è disponibile la classificazione aggiornata, firmata dal Responsabile Sanitario Norma CEI (ambiente ordinario, locale ad uso
(1) Pubblicata nella G.U.C.E. 5 giugno 1992, n. L 154. Entrata in vigore il 22 maggio 1992.
 Pubblicata nella G.U.C.E. 5 giugno 1992, n. L 154. Entrata in vigore il 22 maggio 1992.") Direttiva del 30-4-1992 n. 32 Direttiva del Consiglio recante settima modifica della direttiva 67/548/CEE concernente il ravvicinamento delle disposizioni legislative, regolamentari e amministrative relative
Direttiva del 30-4-1992 n. 32 Direttiva del Consiglio recante settima modifica della direttiva 67/548/CEE concernente il ravvicinamento delle disposizioni legislative, regolamentari e amministrative relative
Roma, 4 Giugno OGGETTO: Decreto Legislativo , n. 46 Nuovi adempimenti previsti per i fabbricanti di dispostivi medici su misura.
 Ministero della Salute DIPARTIMENTO DELL INNOVAZIONE DIREZIONE GENERALE DEI FARMACI E DISPOSITIVI MEDICI Ufficio III - Dispositivi Medici Viale Giorgio Ribotta, 5-00144 Roma DGFDM.III/P/22234/I.5.h.e.1
Ministero della Salute DIPARTIMENTO DELL INNOVAZIONE DIREZIONE GENERALE DEI FARMACI E DISPOSITIVI MEDICI Ufficio III - Dispositivi Medici Viale Giorgio Ribotta, 5-00144 Roma DGFDM.III/P/22234/I.5.h.e.1
Gazzetta ufficiale n. L 169 del 12/07/1993
 TESTO COORDINATO 93/42 CON MODIFICHE O AGGIUNTE INTRODOTTE DA: - Direttiva 2007/47/CE - DIRETTIVA 2001/104/CE - Direttiva 2000/70/CE - Direttiva 98/79/CE DIRETTIVA 93/42/CEE DEL CONSIGLIO (14/06/1993)
TESTO COORDINATO 93/42 CON MODIFICHE O AGGIUNTE INTRODOTTE DA: - Direttiva 2007/47/CE - DIRETTIVA 2001/104/CE - Direttiva 2000/70/CE - Direttiva 98/79/CE DIRETTIVA 93/42/CEE DEL CONSIGLIO (14/06/1993)