Banche dati di sequenze biologiche: interrogazione e ricerca di omologia
|
|
|
- Isabella Bertini
- 5 anni fa
- Visualizzazioni
Transcript
1 Banche dati di sequenze biologiche: interrogazione e ricerca di omologia
2 Raccolte di dati biologici Libri Margaret Dayhoff, collezione di proteine (NBRF) Floppy disk PDB Strutture di macromolecole Cd-rom EMBL (Heidelberg) acidi nucleici 1982 GenBank (NCBI) acidi nucleici Internet EST (Expressed Sequence Tags) WWW Genomi Trascrittomi Oggi: migliaia di collezioni: funzioni, famiglie di proteine, motivi, vie metaboliche...
3 Definizione di banca dati :una collezione di informazioni organizzata in modo che un programma al computer possa velocemente accedere a determinate porzioni di dati Data bank (Banca dati) Collezione di dati Database (Base di dati) Collezione di dati + software per accedervi
4 Struttura Struttura delle delle banche banche dati dati Diagramma della struttura di un database. Un record contiene le informazioni relative ad un dato elemento (entry nei database di sequenza) le cui caratteristiche sono descritte dai fields (campi).
5 Banche dati biologiche Primarie Informazione acquisita direttamente Acidi nucleici Sequenze espresse (EST) Proteine Strutture Secondarie Organizzano informazioni presenti in altre banche dati Famiglie di proteine Famiglie di strutture Promotori
: GenBank (americana) EMBL (europea) DDBJ")
6 Banche dati primarie: acidi nucleici Tre consorzi che scambiano informazioni (International Nucleotide Sequence Database Collaboration): GenBank (americana) EMBL (europea) DDBJ (giapponese)
7 Acidi nucleici: Release e aggiornamenti Una Release in cui la banca dati viene congelata ad una certa data Genetic Sequence Data Bank October NCBI-GenBank Flat File Release Distribution Release Notes loci, bases, from reported sequences This document describes the format and content of the flat files that comprise releases of the GenBank database. If you have any questions or comments about GenBank or this document, please contact NCBI via at info@ncbi.nlm.nih.gov or: + Aggiornamenti quotidiani: Es: GenBank_new, EMBL_new
8 Acidi nucleici: raccolta informazioni Inizialmente informazioni prese dalla letteratura Ora sottomesse direttamente dagli autori. La sottomissione della sequenza alle banche dati è condizione essenziale per pubblicare sulle principali riviste. I dati sono di solito segretati fino alla pubblicazione (entries hold until published) L autore ha il controllo completo della sequenza sottomessa, di conseguenza: solo l autore può modificare l informazione del proprio record, altri non possono correggere l informazione presente anche se questa è chiaramente errata la possibilità di trovare informazioni dipende da quanto accuratamente è stata descritta dall autore

9 Incremento dei dati di sequenza Vs diminuizione dei costi Sequencing costs have dropped several orders of magnitude, from $10 per finished base in 1990 to today's cost, which are estimated at about 5 or 6 cents per base for finished sequence and about 2 to 4 cents for draft sequence. The Scientist 17, 2003

10 Acidi nucleici: quantità di informazione Entries Bases Species Homo sapiens Mus musculus Drosophila melanogaster Rattus norvegicus Oryza sativa Arabidopsis thaliana Caenorhabditis elegans Tetraodon nigroviridis Bos taurus Glycine max Danio rerio Lycopersicon esculentum Medicago truncatula Entamoeba histolytica Xenopus laevis Chlamydomonas reinhardtii Zea mays Strongylocentrotus purpuratus Sus scrofa Trypanosoma brucei
11 Banche dati primarie: EST Una banca dati di sequenze espresse: dbest (Expressed sequence Tag) Le EST sono sequenze relative a piccole porzioni (circa 500 basi) di un mrna, ottenute per sequenziamento parziale di un clone a cdna. il sequenziamento automatico e a singolo passo utilizza primers sul vettore contenente l'inserto. Le sequenze nella banca dati EST corrispondono quindi alle porzioni 5' e 3' terminali del gene.
12 EST: quantità di informazione dbest release Summary by Organism - October 26, Number of public entries: 9,372,718 Homo sapiens (human) 3,859,807 Mus musculus + domesticus (mouse) 2,328,188 Rattus sp. (rat) 317,076 Drosophila melanogaster (fruit fly) 255,456 Glycine max (soybean) 208,186 Bos taurus (cattle) 193,313 Danio rerio (zebrafish) 155,077 Lycopersicon esculentum (tomato) 141,687 Medicago truncatula (barrel medic) 137,588 Caenorhabditis elegans (nematode) 135,203 Xenopus laevis (African clawed frog) 118,996 Arabidopsis thaliana (thale cress) 113,330 Chlamydomonas reinhardtii 111,958 Zea mays (maize) 108,392 Poco più del 50% dei geni umani sequenziati dal genoma ha un corrispondente nelle EST. Le EST rappresentano principalmente il 3' (65%) o il 5' (26%). dei trascritti. Solo nell'11% dei casi le EST 'unite' costituiscono la sequenza completa di un trascritto. Molte EST sono prodotte da company biotech e disponibili a pagamento. Incyte Genomics ha un database privato di 6 milioni di EST e brevetti su diverse migliaia di sequenze.

13 EST: I.M.A.G.E consortium FEATURES source Location/Qualifiers /organism="homo sapiens" /db_xref="taxon:9606" /clone="image:69864" Se il clone da cui deriva la EST appartiene al consorzio I.M.A.G.E è possibile ottenerlo ~ gratuitamente da vari distributori
14 Banche dati primarie: proteine Due consorzi che non scambiano informazioni: SwissProt (europea) PIR (americana) Sequenze di proteine determinate per sequenziamento diretto (in minima parte) Sequenze ricavate dalla traduzione di sequenze codificanti di DNA, di solito annotate e commentate dai curatori della banca Altre banche dati di proteine derivano dalla traduzione di GenBank e EMBL GenPep (GenBank cds) TREMBL(EMBL cds) Sono peggio annotate di SwissProt e Pir, ma più complete
15 Proteine: quantità di informazione Frequency Species Homo sapiens (Human) Mus musculus (Mouse) 9454 Arabidopsis thaliana 7550 Rattus norvegicus (Rat) 6579 Saccharomyces cerevisiae 5792 Bos taurus (Bovine) 4976 Schizosaccharomyces pombe 4429 Escherichia coli 4254 Bacillus subtilis 4253 Dictyostelium discoideum 3306 Caenorhabditis elegans 3273 Xenopus laevis 3090 Drosophila melanogaster 2683 Danio rerio (Zebrafish) 2547 Oryza sativa subsp. japonica 2210 Pongo abelii (Sumatran orangutan) 2179 Gallus gallus (Chicken)
16 Distribuzione Distribuzione della della lunghezze lunghezze delle delle sequenze sequenze proteiche proteiche

17 Distribuzione Distribuzione delle delle frequenze frequenze degli degli amino amino acidi acidi Legend: gray = aliphatic, red = acidic, green = small hydroxy, blue = basic, black = aromatic, white = amide, yellow = sulfur
18 Formato del record: Flat File format Chiave del campo Valore del campo header : testo con le informazioni ( annotazioni ) sulla sequenza Sequenza, memorizzata dal 5 al 3
19 GenBank/DDBJ entry
20 EMBL entry [resto della sequenza omesso]

21 DDBJ/EMBL/GenBank Feature table
22 SwissProt entry ID AC DT DT DT DE GN OS OC OC OX RN RP RX RA RA RT RT RL RP RC CC CC CC CC CC CC CC CC CC CC CC CC CC CC CC RASK_HUMAN STANDARD; PRT; 189 AA. P01116; 21-JUL-1986 (Rel. 01, Created) 21-JUL-1986 (Rel. 01, Last sequence update) 10-OCT-2003 (Rel. 42, Last annotation update) Transforming protein p21a (K-Ras 2A) (Ki-Ras) (c-k-ras). KRAS2 OR RASK2. Homo sapiens (Human). Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo. NCBI_TaxID=9606; [1] SEQUENCE FROM N.A. MEDLINE= ; PubMed= ; [NCBI, ExPASy, EBI, Israel, Japan] McGrath J.P., Capon D.J., Smith D.H., Chen E.Y., Seeburg P.H., Goeddel D.V., Levinson A.D.; "Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene."; Nature 304: (1983). SEQUENCE FROM N.A. TISSUE=Colon carcinoma, and Lung; -!- ALTERNATIVE PRODUCTS: Event=Alternative splicing; Named isoforms=2; Comment=Isoforms differ in the C-terminal region which is encoded by two alternative exons (IVA and IVB); Name=2A; IsoId=P ; Sequence=Displayed; Name=2B; IsoId=P ; Sequence=External; -!- DISEASE: KRAS2 mutations are involved in tumor formation. -!- MISCELLANEOUS: The mammalian ras gene family consists of the Harvey and Kirsten ras genes (c-hras1 and c-kras2), an inactive pseudogene of each (c-hras2 and c-kras1) and the N-ras gene. -!- SIMILARITY: Belongs to the small GTPase superfamily. Ras family. -!- DATABASE: NAME=Atlas Genet. Cytogenet. Oncol. Haematol.;
23 SwissProt entry (continua) DR DR DR DR DR DR DR KW KW FT FT FT FT FT FT FT FT FT FT FT FT FT FT FT SQ BLOCKS; P ProtoNet; P ProtoMap; P PRESAGE; P DIP; P ModBase; P SWISS-2DPAGE; GET REGION ON 2D PAGE. Proto-oncogene; GTP-binding; Prenylation; Palmitate; Lipoprotein; Alternative splicing; Disease mutation. NP_BIND GTP. NP_BIND GTP. NP_BIND GTP. DOMAIN Effector region. DOMAIN HYPERVARIABLE REGION. LIPID S-palmitoyl cysteine. LIPID S-farnesyl cysteine. VARIANT G -> C (in lung carcinoma). /FTId=VAR_ VARIANT G -> V (in colon carcinoma). /FTId=VAR_ VARIANT Q -> H (in lung carcinoma PR310 and pancreas T3M-4). /FTId=VAR_ MUTAGEN R->A: LOSS OF GTP-BINDING ACTIVITY. SEQUENCE 189 AA; MW; B2E11C2C81 CRC64; MTEYKLVVVG AGGVGKSALT IQLIQNHFVD EYDPTIEDSY RKQVVIDGET CLLDILDTAG QEEYSAMRDQ YMRTGEGFLC VFAINNTKSF EDIHHYREQI KRVKDSEDVP MVLVGNKCDL PSRTVDTKQA QDLARSYGIP FIETSAKTRQ RVEDAFYTLV REIRQYRLKK ISKEEKTPGC VKIKKCIIM //
24 EST entry LOCUS H bp mrna EST 24-NOV-1995 DEFINITION PL_16 Root, Angelo Bolchi Zea mays cdna clone PL_16, mrna sequence. ACCESSION H89388 VERSION H GI: KEYWORDS EST. SOURCE Zea mays. REFERENCE 1 (bases 1 to 338) AUTHORS Ottonello,S. TITLE cdnas from maize JOURNAL Unpublished (1995) COMMENT Contact: Simone Ottonello simone@irisbioc.bio.unipr.it. FEATURES Location/Qualifiers source /organism="zea mays" /db_xref="taxon:4577" /clone="pl_16" /clone_lib="root, Angelo Bolchi" /note="vector: pmosblue; Site_1: EcoRV; mrna was purified from the root of sulfate deprived maize seedlings. cdna was constructed using anchored oligo(dt) primers, and PCR amplified in the presence of the same anchored oligo(dt) primer and random primer (Science 257: ). Amplified cdna fragments were ligated into the pmosblue vector (Amersham)." BASE COUNT 91 a 70 c 64 g 113 t ORIGIN 1 cttgttactc caccaaggct atcatgctaa agaaactgct ttatgcgatc aacgaaggcc 61 aagggtcatt tgatctttcg tgaatctcaa cactaacata ggtattggtc cacctagaaa 121 tctgcgtcat tgttacccag agttagtttc tacctcattc atgtatgaca taggttaaac 181 tcagctctcc ggagtcccac cgaagtttgg agccggtacc tttgggtgtg gatgtctata //
25 Porzioni ordinate di GenBank Unigene: Sequenze di Est e di mrna organizzate in cluster che rappersentano un unico gene putativo. RefSeq: Sequenza di riferimento, definita da annotatori, per una data porzione genomica, mrna o proteina Entrez Gene: Loci genetici con informazioni curate da annotatori, relative a funzioni e fenotipi associati

26 Unigene: informazioni sui trascritti
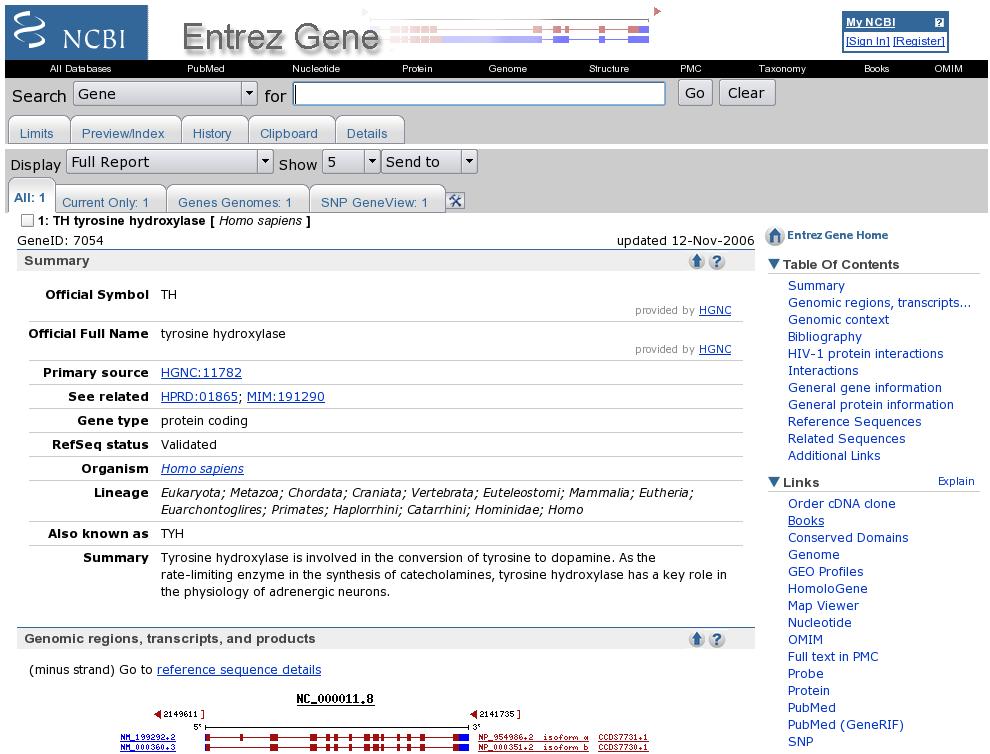
27 Unigene: Profili di espressione Unigine EST Profile di Tirosina idrossilasi (Tirosina ->-> Dopa) Espressa soprattutto nel tessuto nervoso Trascritti per milione (TPM) EST gene / EST totali nel pool
28

29 Dal gene al fenotipo. Online Mendelian Inheritance in Man

30 Dal gene al genoma

31 Dal gene al genoma: analisi del contesto genomico Complete genome > Deinococcus radiodurans Proteina ipotetica Urato ossidasi
32 #1 L ago nel pagliaio Si cerca di solito una sequenza specifica Il numero delle sequenze nella banca dati cresce in modo esponenziale Il numero delle sequenze indesiderate è, in modo crescente, >> delle sequenze desiderate Risultato: trovare quello che si cerca è sempre più difficile
33 #2 Nomenclatura non standard Quando si cercano informazioni in banca dati si dipende dal modo in cui l autore ha deciso di descrivere l informazione. Il modo per designare anche i geni più noti (come il 16s RNA) non è uniforme
 AUTHORS TITLE 'PRODUCTION OF CHIMERIC ANTIBODIES' JOURNAL Patent: WO 8601533-A 3 13-MAR-1986; STANDARD full automatic BASE COUNT 3a 2c 0g 1t ORIGIN 1 cactaa // Sei")
34 #3 Errori e stranezze LOCUS A bp DNA PAT 29-JAN-1993 DEFINITION Nucleotide sequence 3 from patent WO ACCESSION A00674 KEYWORDS. SOURCE Unknown ORGANISM Unknown Unclassified. REFERENCE 1 (bases 1 to 6) AUTHORS TITLE 'PRODUCTION OF CHIMERIC ANTIBODIES' JOURNAL Patent: WO A 3 13-MAR-1986; STANDARD full automatic BASE COUNT 3a 2c 0g 1t ORIGIN 1 cactaa // Sei nucleotidi brevettati di origine sconosciuta transciption, 26sequenze
35 Interrogazione delle banche dati Interrogare una banca dati significa fare una ricerca testuale nella porzione header dei record, contenente le informazioni di testo sulla sequenza. Le banche dati si possono interrogare singolarmente (di solito con il browser attraverso interfacce WWW) oppure con sistemi che consentono di interrogare più banche dati con una sola interfaccia Entrez (NCBI) SRS (Sequence Retrieval System) E importante usare questi sistemi propriamente perché ottenere esattamente l informazione che si cerca interrogando le banche dati di sequenze è difficile.
.")
36 Sistemi di interrogazione: Entrez Entrez è un sistema per cercare e recuperare le informazioni contenute nelle banche dati presenti all NCBI (National Center for Biotechnology Information) e all NLM (National Library of Medicine). Gruppo di database limita la ricerca a in cui effettuare la particolari campi ricerca Rivede le ricerche effettuate per combinarle con operatori logici Le banche dati includono sequenze nucleotidiche e proteiche, strutture molecolari, genomi completi ed informazioni bibliografiche contenute in PubMed (MEDLINE). Il client per l interrogazione può essere il browser o un programma a linea di comando in UNIX (Clever)


37 Sistemi di interrogazione: SRS SRS (Sequence Retrieval System) è un sistema creato all EMBL per interrogare simultaneamente diverse banche dati attraverso un interfaccia comune. Vi sono diversi server SRS pubblici, ciascuno con un particolare subset di banche dati disponibili. Il client per l interrogazione può essere il browser o un programma a linea di comando in UNIX (Getz)

38 Banche dati genomiche

39 Banche dati genomiche: whitehead

40 Museo del genoma
41 Ricerca Ricerca di di omologia omologia in in banca banca dati dati RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti alla data sequenza, ordinate per significatività
42 Ricerca Ricerca in in banca banca dati dati >AAAA acgctaggctagctggatcggggatcggat aggctcggatcgggatttgagtctagggatg >BBBB gctagctggatcggggatcggat ggatcgggatttgagtctagggatg >query >CCCC cgctaggatagctggatcggggatcggat ggctcggatcgggatttgagtctagggatg acgctaggctagctggatcggggatcggat acgctaggctagctggatcggggatcggat acgctaggctagctggatcggggatcggat ccgctaggctagccatcggggatcggat acgctaggctagctggatcggggaaaa 1 Filtro statistico 2 >EEEEE cggctcggatcgggatttgagtctag ccgctaggctagcc... >DDDD acgctaaaaggctagcatcgggga... >DDDD acgctaaaaggctagcatcggggatcggat >EEEEE cggctcggatcgggatttgagtctagggatg ccgctaggctagccatcggggatcggat acgctaggctagctggatcgggg n >AAAA acgctaggctagctggatcggg gatcggat... >FFFFF cggctcggatcgggatttgagtctagggatg ccgctaggctagccatcggggatcggat acgctaggctagctggatcgggg
43 Metodi Metodi per per la la ricerca ricerca in in banca banca dati dati Un algoritmo ottimale di programmazione dinamica ha un tempo di esecuzione proporzionale a N x M (lunghezza della sequenza per dimensione della banca dati). In computer molto potenti (paralleli) è possibile usare un algoritmo di programmazione dinamica per una ricerca più accurata in tempi brevi. Allineamento ottimale SSEARCH (Smith-Waterman) Gli algoritmi euristici usano scorciatoie che abbreviano anche di 50 volte il tempo di esecuzione senza garantire un allineamento ottimale Allineamento euristico FASTA BLAST
44 SSEARCH SSEARCH Utilizza un algoritmo completo di programmazione dinamica. Equivale ad un allineamento locale (Smith e Waterman) tra la query sequence e ciascuna sequenza della banca dati. Meno veloce di BLAST e FASTA ma garantisce un allineamento ottimale e la massima accuratezza
45 Metodi euristici: confronto tra indici query sequence H A R F Y A A Q I V L Ktup = 1 Indice (Lookup table) A 2, 6, 7 F 4 H 1 I 9 L 11 Q 8 R 3 V 10 Y 5 Database sequence V D M A A Q I A offsets Offset vector
46 Il confronto tra indici trova rapidamente segmenti simili H A R F Y A A Q I V L V D M A A Q I A Offset: Segmenti con il medesimo offset sono simili
47 Segmenti simili sono rappresentati come diagonali con il medesimo offset H A R F Y A A Q I V L V D M A A Q I A
48 FASTA Pearson & Lipmann, PNAS 1988 K-tuple = lungezza delle parole nell'indice k-tuple k-tuple k-tuple = 4 sensibilità velocità sensibilità velocità Valori standard: k-tuple=2 (proteine) k-tuple=6 (nucleotidi)
49 FASTA FASTA (schema) (schema) Rappresenta la query sequence e le sequenze nella banca dati con una tabella di parole (Lookup table). La lunghezza della parola è definita dal valore ktuple. Confronta le posizioni delle parole e identifica le regioni di match (diagonali). Le migliori diagonali sono estese per trovare i match più lunghi senza gap. I segmenti diagonali migliori vengono uniti con gap (se il punteggio complessivo è migliore tenendo conto della penalità per i gap) La migliore regione viene riallineata con programmazione dinamica, limitando l allineamento a una banda della matrice
50 BLAST Altschul et al JMB 1990 Basic Local Alignment Tool Words (parole indice) T (punteggio minimo per parola) W T W T sensibilità velocità sensibilità velocità >query AGPDPATA AGP GPD PDP DPA PAT ATA words + PEP, EPA, DPG, Neighbourhood words La lunghezza delle words è definita dal parametro W. Il punteggio che devono raggiungere per essere considerate è definito dal parametro T.
51 BLAST BLAST (schema) (schema) Per la query sequence indicizza tutte le parole (words) di una lunghezza data (2 o 3 per le proteine, 11 per gli acidi nucleici) compilando una lista che include tutte le parole simili con un dato punteggio di match rispetto ad una soglia. Per ogni match con la banca dati, estende i segmenti a maggior punteggio (High Scoring Pairs, HSP) fino a quando lo score totale aumenta. Nella versione originale non ammetteva gap, le nuove versioni (gapped-blast) producono allineamenti con gap
52 Calcolo della probabilità casuali di un allineamento La funzione di probabilità degli score casuali in un allineamento segue la distribuzione dei valori estremi (EVD) la probabilità di ottenere per caso almeno un segmento con score S>x è uguale a: P S x = 1 e E Dove E (Expect) è in numero di segmenti attesi raggiungere un certo score per effetto del caso
53 Calcolo del numero di segmenti attesi (E) Il numero di segmenti attesi con un punteggio x>s per effetto del caso in confronti a coppie è calcolabile dalla distribuzione dei valori estremi (EVD) e dipende: 1) dal punteggio 2) dalla dimensione delle sequenze 3) dal sistema di punteggio usato (matrice e penalità) E = Kmne λs S è lo score dell'allineamento m e n sono le dimensioni delle sequenze confrontate. Nella ricerca di omologia il termine tiene conto delle dimensioni della banca dati K e λ dipendono dal tipo di matrice usata e dalle penalità assegnate ai gap e dalla composizione delle sequenze. Questi parametri possono essere stimati con fitting osservando la distribuzione degli score dei segmenti casuali. In BLAST sono precalcolati per ogni matrice e penalità di gap In FASTA sono calcolati sulla distribuzione dei punteggi della banca dati
54 Significato di E nella ricerca di omologia Il valore di E è usato come misura della significatività dell'allineamento Un allineamento è significativo se è improbabile che si possa ottenere per effetto del caso P S x = 1 e E Se E è molto piccolo, ha il significato di una probabilità. Per E<<1, E P Negli altri casi ha il significato di numero di segmenti casuali attesi. Per E>>1, P 1
55 FASTA FASTA Output Output < > :=== 0:= one = represents 1345 library sequences 1:* 17:* 178:* 1084:*= 4192:===* 11369:========*== 23349:=================* 38587:============================*= 53825:========================================*= 65795:================================================*==== 72578:=====================================================*====== 73923:======================================================*==== 70772:====================================================*= 64580:================================================* 56777:========================================= * Statistiche ottenute dai punteggi con la banca dati. Fitting sulla EVD per determinare i valori dei parametri λ e k 241:*:=====================*==== 186:*:================*======== 144:*:=============*====== 112:*:==========*===== residues in sequences 86:*:=======*==== statistics extrapolated from to sequences 67:*:======*== Expectation_n fit: rho(ln(x))= / ; mu= / :*:====*==== mean_var= / , 0's: 530 Z-trim: 573 B-trim: 974 in 2/62 40:*:===*==== Lambda= :*:==*===== Kolmogorov-Smirnov statistic: (N=29) at 48 24:*:==*=== 19:*:=*=== 14:*:=*======================================
56 FASTA FASTA output output 22 score Lista significatività The best scores are: gi ref NP_ High mobility group (HMG)-like protein; gi emb CAA (X59863) a xenopus upstream binding factor gi pir S17196 transcription factor UBF2 - African clawed fro gi sp P25980 UBF2_XENLA NUCLEOLAR TRANSCRIPTION FACTOR 2 (UPS gi sp P40626 HMGB_TETTH HIGH-MOBILITY-GROUP PROTEIN B gi 1023 gi pir T38936 non-histone chromosomal protein high mobility gi pir S47596 HMG1-like protein - fruit fly (Drosophila mel gi sp P41848 SSRP_CAEEL PROBABLE STRUCTURE-SPECIFIC RECOGNIT gi ref NP_ high mobility group protein (HMG1), put ( ( ( ( ( ( ( ( ( opt bits E(787946) 246) e ) ) ) ) ) ) ) ) gi pir JC5112 ribosomal transcription factor UBF1 - Chinese gi gb AAB (L42570) putative [Cricetulus griseus] gi pir A24019 nonhistone chromosomal protein HMG-T - trout (f gi dbj BAB (AK004961) putative [Mus musculus] gi pir B40439 UBF transcription factor, short form - rat gi ref XP_ hypothetical protein XP_ [Homo sa gi ref XP_ high-mobility group 20A [Homo sapiens] gi sp Q04931 SSRP_RAT STRUCTURE-SPECIFIC RECOGNITION PROTEIN gi ref NP_ high mobility group 20A [Mus musculus] ( ( ( ( ( ( ( ( ( 764) 764) 172) 752) 727) 727) 313) 561) 346) Allineamenti >>gi ref NP_ High mobility group (HMG)-like protein; Hmo1p (246 aa) QUERY MTTDPSVKLKSAKDSLVSSLFELSKAANQTASSIVDFYNAIGDDEEEKIEAFTTLTESLQTLTSGVNHLHGISSELVNPI :::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: gi 632 MTTDPSVKLKSAKDSLVSSLFELSKAANQTASSIVDFYNAIGDDEEEKIEAFTTLTESLQTLTSGVNHLHGISSELVNPI
57 BLAST BLAST Output Output Sequences producing significant alignments: Score (bits) E Value gi ref NP_ High mobility group (HMG)-like... gi pir T12113 transcription factor - fava bean >gi... gi sp Q09390 YR44_CAEEL HYPOTHETICAL 23.8 KD PROTEI... gi gb AAK U22831_8 (U22831) Hypothetical pr... gi ref NP_ structure specific recognition... gi pir T43009 HMG protein Caenorhabditis el... gi gb AAK U22831_9 (U22831) Hypothetical pr... gi dbj BAB (AK017716) putative [Mus musculus] gi ref NP_ high mobility group 20A [Mus m... gi ref NP_ high-mobility group 20A [Homo s... gi pir JC6179 dorsal switch protein 1 - fruit fly... gi pir S50068 nonhistone chromosomal protein HMG1-... gi sp P25980 UBF2_XENLA NUCLEOLAR TRANSCRIPTION FACT... gi emb CAA (X59863) a xenopus upstream bindi... gi emb CAA (X81456) unnamed protein product e-90 1e-05 8e-05 1e-04 1e-04 1e-04 1e-04 2e-04 3e-04 3e-04 3e-04 3e-04 4e-04 4e-04 4e-04 gi sp Q91731 SX11_XENLA TRANSCRIPTION FACTOR SOX gi ref XP_ hypothetical protein XP_ gi pdb 1AAB Nmr Structure Of Rat Hmg1 Hmga Frag... gi dbj BAB (AK004857) putative [Mus musculus] gi pdb 1HME High Mobility Group Protein Fragment... gi pir T03375 high mobility group protein HMGd gi gb AAK (AC024859) Hypothetical protein
58 BLAST BLAST Output Output 22 Allineamenti >gi ref NP_ High mobility group (HMG)-like protein; Hmo1p [Saccharomyces Length = 246 Score = 332 bits (850), Expect = 3e-90 Identities = 191/220 (86%), Positives = 191/220 (86%) Query: 1 Sbjct: 1 Query: 61 Sbjct: MTTDPSVKLKSAKDSLVSSLFELSKAANQTASSIVDFYNAIGDDEEEKIEAFXXXXXXXX 60 MTTDPSVKLKSAKDSLVSSLFELSKAANQTASSIVDFYNAIGDDEEEKIEAF MTTDPSVKLKSAKDSLVSSLFELSKAANQTASSIVDFYNAIGDDEEEKIEAFTTLTESLQ 60 XXXXGVNHLHGISSELVNXXXXXXXXXXXXXXXXXRRKIERDPNAPKKPLTVFFAYSAYV 120 GVNHLHGISSELVN RRKIERDPNAPKKPLTVFFAYSAYV TLTSGVNHLHGISSELVNPIDDDKDAIIAAPVKAVRRKIERDPNAPKKPLTVFFAYSAYV 120 Lambda Gapped Lambda K Statistiche basate su valori di λ e k pre-calcolati per determinate matrici, composizione in residui e penalità per gap H K H Matrix: BLOSUM62 Gap Penalties: Existence: 11, Extension: 1
59 Punteggi Punteggi normalizzati normalizzati (Bits (Bits score) score) Lo score di un confronto nella ricerca di omologia viene espresso sia come raw score, sia come score normalizzato (bits score). La relazione che lega S' ad E diventa:
60 Regioni a bassa complessità Le regioni a bassa complessità hanno una diversa statistica dei punteggi casuali poiché le probabilità di match casuali sono molto più alte La sequenza LRVSQQQQQQQQQQQAAPPPPPPPPPPKDFCV Avrà molte più provabilità di avere falsi match con altre sequenze con simile composizione aminoacidica Appositi programmi (tra cui SEG) identificano tali regioni nelle sequenze e le mascherano prima del confronto. La sequenza verrà quindi trasformata in: LRVSXXXXXXXXXXXXXXXXXXXXXXKDFCV (Ora scritta come LRVSQQQQQQQQQQQAAPPPPPPPPPPKDFCV) Il filtro può essere attivo a default in BLAST. In alcuni casi, quando si vuole ricercare altre sequenze con simili ripetizioni, è opportuno disattivare il filtro

61 Output Output grafico grafico di di blast blast
62 Significatività e dimensioni delle bancadati La significatività dei punteggi di somiglianza decresce al crescere delle dimensioni della banca dati: Il punteggio del confronto tra due sequenze è costante Il numero delle sequenze nella banca dati cresce in modo esponenziale Il numero nelle sequenze non omologhe >> di quelle omologhe E necessaria una sensibilità sempre maggiore per identificare le sequenze omologhe
63 II programmi programmi della della famiglia famiglia BLAST BLAST ee FASTA FASTA P ro g ra m m a Q u e r yb a n c a d a tit ip o d i c o n fr o n to B L A S TF A S T A b la s tn fa s ta 3 nuc nuc nuc / nuc b la s tp fa s ta 3 aa aa aa / aa b la s tx fa s tx3 fa s ty3 nuc aa aa / aa tb la s tn tfa s tx3 tfa s ty3 aa nuc a a /a a tb la s tx nuc nuc aa / aa
64 E(DNA) vs E(Proteine) Valori attesi con la sequenza di DNA [Score, E(DNA)] e la sequenza proteica [E(prot)] per una ricerca in bancadati effettuata con la glutatione transferasi (GST) di drosofila. La ricerca che utilizza la sequenza proteica fornisce risultati più significativi ed è in grado di individuare omologhi della GST al di fuori del gruppo degli insetti
65 Misure dell accuratezza della ricerca SENSIBILITA o COPERTURA [ VP / (VP + FN) ] := Rapporto tra il numero delle sequenze trovate effettivamente omologhe (veri positivi) e il numero totale delle sequenze omologhe (veri positivi + falsi negativi) SELETTIVITA [ VP / (VP + FP) ] := Rapporto tra il numero delle sequenze trovate effettivamente omologhe (veri positivi) e il numero totale delle sequenze trovate (veri positivi + falsi positivi) ERRORE [ FP / (VP + FP) ] := Rapporto tra le sequenze trovate ma non omologhe (falsi positivi) e il numero totale delle sequenze trovate (veri positivi + falsi positivi)
66 Compromesso tra sensibilità e selettività Sequenze non appartenenti alla famiglia Sequenze appartenenti alla famiglia Se si considera la distribuzione degli score delle sequenze imparentate rispetto ai falsi positivi, molto difficilmente si hanno due curve perfettamente separate. Una soglia stringente consente di eliminare i falsi positivi ma ha anche l effetto di escludere alcuni veri positivi. Viceversa, una soglia tollerante comprende tutti i membri della famiglia ma anche molti falsi positivi. Un compromesso spesso usato è scegliere la soglia all intersezione delle curve.
67 Copertura vs Errore 100% soglia=30 Errore. % falsi positivi sul totale soglia=20 soglia=10 Diverse soglie di punteggo Sensibilità (Copertura). Due metodi. Quello tratteggiato è peggiore 100% % veri positivi sul totale
68 Accuratezza dei metodi di ricerca Brenner et al PNAS 1998 Prestazioni dei programmi di ricerca di omologia nelle superfamiglie di SCOP (omologhi strutturali) con identità di sequenza < del 40% SSEARCH > FASTA > BLAST METODO SSEARCH E-values FASTA ktup = 1 E-values FASTA ktup = 2 E-values WU-BLAST2 E-values BLAST E-values TEMPO (s) COPERTURA 1% Err
69 Argomenti pratici nella ricerca in banca dati Sapere dove cercare! (usare la banca dati più completa. Notare che alcune sezioni dbest, HTGS, Patent sono tenute separate dalla banca dati principale) Usare le matrici e le penalità dei i gap opportuni Preferire una ricerca con la traduzione in amino acidi se la query sequence è codificante Utilizzare SSEARCH per il massimo dell accuratezza Utilizzare FASTA (ktup=1) per una ricerca nucleotidica In BLAST fare attenzione all opzione filtro Esaminare con attenzione l allineamento e la feature table delle sequenze simili prima di fare deduzioni su omologia e funzione Ricordarsi dei genomi completi. Prestate attenzione anche a quello che non c è.
Ricerca di omologia di sequenza
 Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Banche dati di sequenze biologiche: Organizzazione e Interrogazione
 Banche dati di sequenze biologiche: Organizzazione e Interrogazione Raccolte di dati biologici Libri - 1960 Margaret Dayhoff, collezione di proteine (NBRF) Floppy disk Cd rom - 1977 PDB Strutture di macromolecole
Banche dati di sequenze biologiche: Organizzazione e Interrogazione Raccolte di dati biologici Libri - 1960 Margaret Dayhoff, collezione di proteine (NBRF) Floppy disk Cd rom - 1977 PDB Strutture di macromolecole
 http://biochimica.unipr.it/biocomp/ab/bioinformatica/index.html Banche dati di sequenze biologiche: Organizzazione e Interrogazione Raccolte di dati biologici Libri - 1960 Margaret Dayhoff, sequenze di
http://biochimica.unipr.it/biocomp/ab/bioinformatica/index.html Banche dati di sequenze biologiche: Organizzazione e Interrogazione Raccolte di dati biologici Libri - 1960 Margaret Dayhoff, sequenze di
Internet web: >8,000,000,000 pagine
 Internet web: >8,000,000,000 pagine Merck Index: >10.000 monografie su composti chimici Uric Acid Ammonia is a universal participant in amino acid synthesis and degradation, but its accumulation has toxic
Internet web: >8,000,000,000 pagine Merck Index: >10.000 monografie su composti chimici Uric Acid Ammonia is a universal participant in amino acid synthesis and degradation, but its accumulation has toxic
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Lezione 8. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 6. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 6 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 6 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Z-score. lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random
/ deviazione standard random") Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Bioinformatica ed applicazioni di bioinformatica strutturale!
 Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti Esercitazione 7 2.
 Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti patti@di.unito.it Esercitazione 7 1 Info&Bio Bio@Lab Allineamento di sequenze Esercitazione 7 2 1 Es2: Allineamento
Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti patti@di.unito.it Esercitazione 7 1 Info&Bio Bio@Lab Allineamento di sequenze Esercitazione 7 2 1 Es2: Allineamento
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
Il progetto Genoma Umano è iniziato nel E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA.
 Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Laboratorio di Bioinformatica I. Parte 1. Dott. Sergio Marin Vargas (2014 / 2015)
") Laboratorio di Bioinformatica I Banche dati Parte 1 Dott. Sergio Marin Vargas (2014 / 2015) Introduzione a NCBI National Center for Biotechnology Information (NCBI) http://www.ncbi.nlm.nih.gov/ NCBI Databases
Laboratorio di Bioinformatica I Banche dati Parte 1 Dott. Sergio Marin Vargas (2014 / 2015) Introduzione a NCBI National Center for Biotechnology Information (NCBI) http://www.ncbi.nlm.nih.gov/ NCBI Databases
Banche Dati Primarie di Biosequenze
 Descrizione Ie banche dati primarie delle sequenze nucleotidiche EMBL, GenBank e DDBJ sono una collezione di sequenze di DNA e RNA che provengono dalla letteratura scientifica e dalle sequenze brevettate.
Descrizione Ie banche dati primarie delle sequenze nucleotidiche EMBL, GenBank e DDBJ sono una collezione di sequenze di DNA e RNA che provengono dalla letteratura scientifica e dalle sequenze brevettate.
Bioinformatica. :studio dei problemi biologici attraverso le metodologie dell'informatica
 Bioinformatica :studio dei problemi biologici attraverso le metodologie dell'informatica Sinomimi: biochimica computazionale, biologia molecolare computazionale Viceversa: Biocomputazione, algoritmi genetici,
Bioinformatica :studio dei problemi biologici attraverso le metodologie dell'informatica Sinomimi: biochimica computazionale, biologia molecolare computazionale Viceversa: Biocomputazione, algoritmi genetici,
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Banche Dati proteiche
 Banche Dati proteiche Un altro grande database è UniProt, The Universal Protein Resource (http://www.uniprot.org/) nel quale sono radunate le sequenze proteiche, e le annotazione delle stesse, ottenute
Banche Dati proteiche Un altro grande database è UniProt, The Universal Protein Resource (http://www.uniprot.org/) nel quale sono radunate le sequenze proteiche, e le annotazione delle stesse, ottenute
Laboratorio di Metodologie e Tecnologie Genetiche ESERCITAZIONE DI BIOINFORMATICA
 Laboratorio di Metodologie e Tecnologie Genetiche ESERCITAZIONE DI BIOINFORMATICA Bioinformatica - Scienza interdisciplinare coinvolgente la biologia, l informatica, la matematica e la statistica per l
Laboratorio di Metodologie e Tecnologie Genetiche ESERCITAZIONE DI BIOINFORMATICA Bioinformatica - Scienza interdisciplinare coinvolgente la biologia, l informatica, la matematica e la statistica per l
Corso di Bioinformatica e analisi dei genomi, docente Silvia Fuselli. Esercizi ricerche in banche dati
 Corso di Bioinformatica e analisi dei genomi, docente Silvia Fuselli Esercizi ricerche in banche dati 1) Nel romanzo fantasy Jurassic Park di Michael Crichton sulla possibilità di clonare i dinosauri,
Corso di Bioinformatica e analisi dei genomi, docente Silvia Fuselli Esercizi ricerche in banche dati 1) Nel romanzo fantasy Jurassic Park di Michael Crichton sulla possibilità di clonare i dinosauri,
Corso di Elementi di Bioinformatica
 Corso di Elementi di Bioinformatica Laurea Triennale in Informatica I dati e le banche dati in Bioinformatica Anno Accademico 2015-2016 Docente del laboratorio: Raffaella Rizzi 1 Il DNA (oggetto biologico)
Corso di Elementi di Bioinformatica Laurea Triennale in Informatica I dati e le banche dati in Bioinformatica Anno Accademico 2015-2016 Docente del laboratorio: Raffaella Rizzi 1 Il DNA (oggetto biologico)
Database biologici (banche di dati biologici)
") 1 Lo sviluppo di tecnologie strumentali sempre più sofisticate ha portato ad una enorme produzione di dati biologici. Per la gestione di questi dati è quindi necessario disporre di potenti sistemi di archiviazione
1 Lo sviluppo di tecnologie strumentali sempre più sofisticate ha portato ad una enorme produzione di dati biologici. Per la gestione di questi dati è quindi necessario disporre di potenti sistemi di archiviazione
Sommario. Presentazione dell opera Ringraziamenti
 Sommario Presentazione dell opera Ringraziamenti XI XII Capitolo 1 Introduzione alla bioinformatica 1 1.1 Cenni introduttivi 1 1.2 Pietre miliari della bioinformatica 2 1.3 Infrastrutture bioinformatiche
Sommario Presentazione dell opera Ringraziamenti XI XII Capitolo 1 Introduzione alla bioinformatica 1 1.1 Cenni introduttivi 1 1.2 Pietre miliari della bioinformatica 2 1.3 Infrastrutture bioinformatiche
Bellini Lara matricola: Tesina di Biologia Molecolare 2
 Bellini Lara matricola: 594736 Tesina di Biologia Molecolare 2 Argomento: Scegli una proteina di Drosophila e trovala in Uniprot.Descrivi le informazioni presenti nel record ed i collegamenti a risorse
Bellini Lara matricola: 594736 Tesina di Biologia Molecolare 2 Argomento: Scegli una proteina di Drosophila e trovala in Uniprot.Descrivi le informazioni presenti nel record ed i collegamenti a risorse
Ricerche con BLAST (Laboratorio)
") Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Metodi euristici di allineamento
 Metodi euristici di allineamento Algoritmi euristici di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca rapida, anche se meno accurata, utilizzando la similarità
Metodi euristici di allineamento Algoritmi euristici di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca rapida, anche se meno accurata, utilizzando la similarità
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
Banche Dati. Docente: Dr. Antinisca DI MARCO
 Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@di.univaq.it La biologia molecolare produce una grande mole di dati che può essere memorizzata in database general-purpose o specialized (es. immunological):
Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@di.univaq.it La biologia molecolare produce una grande mole di dati che può essere memorizzata in database general-purpose o specialized (es. immunological):
GENOMA. Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine CONTENUTO FUNZIONE. Progetti genoma in centinaia di organismi
 GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
Laboratorio di Elementi di Bioinformatica
 Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2015/2016 Parsing di un file in formato EMBL (parte I) Docente del laboratorio: Raffaella Rizzi 1 Esercizio
Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2015/2016 Parsing di un file in formato EMBL (parte I) Docente del laboratorio: Raffaella Rizzi 1 Esercizio
Tesina di Biologia Molecolare II
 MELATO GIULIA 595033 Tesina di Biologia Molecolare II Mostra un albero filogenetico con la relazione tra Uomo, Topo e Ratto. Che banca dati è disponibile per quest'ultimo organismo? Descrivi alcune caratteristiche
MELATO GIULIA 595033 Tesina di Biologia Molecolare II Mostra un albero filogenetico con la relazione tra Uomo, Topo e Ratto. Che banca dati è disponibile per quest'ultimo organismo? Descrivi alcune caratteristiche
Database genomici primari
 Esercitazione di laboratorio di bioinformatica Seconda parte: I principali database genomici e proteomici Slide ricavate dal corso di Laboratorio Integrato di Biologia Computazionale Francesca Cordero
Esercitazione di laboratorio di bioinformatica Seconda parte: I principali database genomici e proteomici Slide ricavate dal corso di Laboratorio Integrato di Biologia Computazionale Francesca Cordero
La ricerca di similarità: i metodi
 La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
31/05/2007. Omologia. Evoluzione: Mutabilità e Selezione Naturale. Similarità. Sequenze omologhe sono sempre simili?
 . 31/05/2007 Evoluzione Molecolare e omologia Evoluzione Molecolare e omologia Evoluzione: Mutabilità e Selezione Naturale Le sequenze degli organismi attuali hanno avuto origine dall evoluzione di sequenze
. 31/05/2007 Evoluzione Molecolare e omologia Evoluzione Molecolare e omologia Evoluzione: Mutabilità e Selezione Naturale Le sequenze degli organismi attuali hanno avuto origine dall evoluzione di sequenze
Laboratorio di Elementi di Bioinformatica
 Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2016/2017 I dati in Bioinformatica Docente del laboratorio: Raffaella Rizzi 1 Il DNA (oggetto biologico)
Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2016/2017 I dati in Bioinformatica Docente del laboratorio: Raffaella Rizzi 1 Il DNA (oggetto biologico)
Laboratorio di Bioinformatica I. Parte 2. Dott. Sergio Marin Vargas (2014 / 2015)
") Laboratorio di Bioinformatica I Banche dati Parte 2 Dott. Sergio Marin Vargas (2014 / 2015) Google Scholar https://scholar.google.it/ E un motore di ricerca di Google, specializzato nella ricerca di articoli
Laboratorio di Bioinformatica I Banche dati Parte 2 Dott. Sergio Marin Vargas (2014 / 2015) Google Scholar https://scholar.google.it/ E un motore di ricerca di Google, specializzato nella ricerca di articoli
InfoBioLab I ENTREZ. ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche
 InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
DataBase Biologici 1
 DataBase Biologici 1 Lo sviluppo di tecnologie strumentali sempre più sofisticate ha portato ad una enorme produzione di dati biologici. Per la gestione di questi dati è quindi necessario disporre di potenti
DataBase Biologici 1 Lo sviluppo di tecnologie strumentali sempre più sofisticate ha portato ad una enorme produzione di dati biologici. Per la gestione di questi dati è quindi necessario disporre di potenti
Il Corso sarà tenuto nei giorni di Lunedì, Mercoledì e Venerdì dalle ore 17 alle ore 19.
 Docente: Prof. Alfredo Ferro Il Corso sarà tenuto nei giorni di Lunedì, Mercoledì e Venerdì dalle ore 17 alle ore 19. Programma del Corso DATA ARGOMENTO 09/03/2011 Introduzione al corso. Slides Panoramica
Docente: Prof. Alfredo Ferro Il Corso sarà tenuto nei giorni di Lunedì, Mercoledì e Venerdì dalle ore 17 alle ore 19. Programma del Corso DATA ARGOMENTO 09/03/2011 Introduzione al corso. Slides Panoramica
Informatica e Bioinformatica: Basi di Dati
 Informatica e Bioinformatica: Date TBD Bioinformatica I costi di sequenziamento e di hardware descrescono vertiginosamente si hanno a disposizione sempre più dati e hardware sempre più potente e meno costoso...
Informatica e Bioinformatica: Date TBD Bioinformatica I costi di sequenziamento e di hardware descrescono vertiginosamente si hanno a disposizione sempre più dati e hardware sempre più potente e meno costoso...
SAGA: sequence alignment by genetic algorithm. ALESSANDRO PIETRELLI Soft Computing
 SAGA: sequence alignment by genetic algorithm ALESSANDRO PIETRELLI Soft Computing Bologna, 25 Maggio 2007 Multi Allineamento di Sequenze (MSAs) Cosa sono? A cosa servono? Come vengono calcolati Multi Allineamento
SAGA: sequence alignment by genetic algorithm ALESSANDRO PIETRELLI Soft Computing Bologna, 25 Maggio 2007 Multi Allineamento di Sequenze (MSAs) Cosa sono? A cosa servono? Come vengono calcolati Multi Allineamento
Bioinformatica. Analisi del genoma
 Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
RELAZIONE di BIOLOGIA MOLECOLARE
 NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
Provate rispondere alle domande, se ci riuscirete, sarete pronti a superare l esame per quanto riguarda la parte di bioinformatica.
 Per aiutarvi ho elaborato (frettolosamente) questi quesiti che dovrebbero aiutarvi ad individuare gli argomenti importanti del corso ed a darvi un idea delle domande che potrebbero esservi poste all esame.
Per aiutarvi ho elaborato (frettolosamente) questi quesiti che dovrebbero aiutarvi ad individuare gli argomenti importanti del corso ed a darvi un idea delle domande che potrebbero esservi poste all esame.
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini)
") Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Programmazione dinamica
 Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
L organizzazione del genoma. Prof. Savino; dispense di Biologia Molecolare, Corso di Laurea in Biotecnologie
 L organizzazione del genoma L organizzazione del genoma Fino ad ora abiamo studiato la regolazione dell espressione genica prendendo come esempio singoli geni dei batteri. Ma quanti geni ci sono in un
L organizzazione del genoma L organizzazione del genoma Fino ad ora abiamo studiato la regolazione dell espressione genica prendendo come esempio singoli geni dei batteri. Ma quanti geni ci sono in un
Esercitazioni Informatiche e Telematiche
 Esercitazioni Informatiche e Telematiche Scuola di Farmacia e Nutraceutica Università Magna Graecia di Catanzaro I Anno, I Semestre, A.A. 2015/2016 Ing. Alessia Sarica 2 Informazioni Docente Ing. Alessia
Esercitazioni Informatiche e Telematiche Scuola di Farmacia e Nutraceutica Università Magna Graecia di Catanzaro I Anno, I Semestre, A.A. 2015/2016 Ing. Alessia Sarica 2 Informazioni Docente Ing. Alessia
Genomica, proteomica, genomica strutturale, banche dati.
 Genomica, proteomica, genomica strutturale, banche dati. Alcune pietre miliari della biologia anno risultato 1866 Mendel scopre i geni 1944 il DNA è il materiale genetico 1951 prima sequenza di una proteina
Genomica, proteomica, genomica strutturale, banche dati. Alcune pietre miliari della biologia anno risultato 1866 Mendel scopre i geni 1944 il DNA è il materiale genetico 1951 prima sequenza di una proteina
Informatica e biotecnologie
 Informatica e biotecnologie Ricerca di informazioni e analisi di sequenze CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Informatica e biotecnologie Strumenti per raccogliere
Informatica e biotecnologie Ricerca di informazioni e analisi di sequenze CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Informatica e biotecnologie Strumenti per raccogliere
Informatica e Bioinformatica A. A
 GQuery (http://www.ncbi.nlm.nih.gov/gquery/) è il punto di partenza per eseguire query su tutti o parte dei database dell NCBI: si basa sul sistema di interrogazione ENTREZ Informatica e Bioinformatica
GQuery (http://www.ncbi.nlm.nih.gov/gquery/) è il punto di partenza per eseguire query su tutti o parte dei database dell NCBI: si basa sul sistema di interrogazione ENTREZ Informatica e Bioinformatica
Database di sequenze. Dati di sequenza. Caratteristiche dei dati della biologia molecolare. I dati ed i problemi della bioinformatica
 I dati ed i problemi della bioinformatica Giorgio Valentini DSI Università degli Studi di Milano 1 Caratteristiche dei dati della biologia molecolare Diverse tipologie di dati bio-molecolari Per ogni tipo
I dati ed i problemi della bioinformatica Giorgio Valentini DSI Università degli Studi di Milano 1 Caratteristiche dei dati della biologia molecolare Diverse tipologie di dati bio-molecolari Per ogni tipo
Programmazione dinamica
 rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Ogni tipo ha il suo alfabeto di riferimento, e metodi specifici, nonché metodi per la conversione da un tipo all altro (trascrizione, traduzione)
") BioPython Descrizione Il progetto BioPython è un associazione di sviluppatori di codice Python liberamente disponibile per bioinformatica La homepage del progetto è http://www.biopython.org Il codice viene
BioPython Descrizione Il progetto BioPython è un associazione di sviluppatori di codice Python liberamente disponibile per bioinformatica La homepage del progetto è http://www.biopython.org Il codice viene
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Modello computazionale per la predizione di siti di legame per fattori di trascrizione
 Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA
 METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA Scopo di questa esercitazione è apprendere l utilizzo di internet per: STUDIO DELLA STRUTTURA E DELLA FUNZIONE DELLE PROTEINE Conoscere i database
METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA Scopo di questa esercitazione è apprendere l utilizzo di internet per: STUDIO DELLA STRUTTURA E DELLA FUNZIONE DELLE PROTEINE Conoscere i database
Principi di biologia
 Principi di biologia Prof.ssa Flavia Frabetti Tecnici di lab. 2009-10 BIOLOGIA è la scienza della vita, che indaga le caratteristiche dei sistemi viventi biologia animale biologia cellulare biologia molecolare
Principi di biologia Prof.ssa Flavia Frabetti Tecnici di lab. 2009-10 BIOLOGIA è la scienza della vita, che indaga le caratteristiche dei sistemi viventi biologia animale biologia cellulare biologia molecolare
Applicazione della biologia molecolare nella valutazione del benessere del cavallo
 UNIVERSITA DEGLI STUDI DI PERUGIA FACOLTA DI MEDICINA VETERINARIA Centro di Studio del Cavallo Sportivo Applicazione della biologia molecolare nella valutazione del benessere del cavallo Andrea Verini
UNIVERSITA DEGLI STUDI DI PERUGIA FACOLTA DI MEDICINA VETERINARIA Centro di Studio del Cavallo Sportivo Applicazione della biologia molecolare nella valutazione del benessere del cavallo Andrea Verini
Banche dati molti dati sulle proteine derivano dalle banche dati primarie
 Banche dati Banche dati Si possono raggruppare in varie categorie in base al tipo di dato biologico che raccolgono e organizzano, ma ce ne sono alcune che sono da considerarsi fondamentali: - banche dati
Banche dati Banche dati Si possono raggruppare in varie categorie in base al tipo di dato biologico che raccolgono e organizzano, ma ce ne sono alcune che sono da considerarsi fondamentali: - banche dati
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Laboratorio di Elementi di Bioinformatica
 Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2016/2017 Formato GTF per annotare un gene Docente del laboratorio: Raffaella Rizzi 1 GTF (Gene Transfer
Laboratorio di Elementi di Bioinformatica Laurea Triennale in Informatica (codice: E3101Q116) AA 2016/2017 Formato GTF per annotare un gene Docente del laboratorio: Raffaella Rizzi 1 GTF (Gene Transfer
Come facciamo ad isolare un gene da un organismo? Utilizziamo una libreria ovvero una collezione dei geni del genoma del cromosoma di un organismo
 Come facciamo ad isolare un gene da un organismo? Utilizziamo una libreria ovvero una collezione dei geni del genoma del cromosoma di un organismo GENOMA di alcuni organismi viventi raffigurato come libri
Come facciamo ad isolare un gene da un organismo? Utilizziamo una libreria ovvero una collezione dei geni del genoma del cromosoma di un organismo GENOMA di alcuni organismi viventi raffigurato come libri
Introduzione alla Genomica
 Laboratorio di Bioinformatica I Introduzione alla Genomica Dott. Sergio Marin Vargas (2014 / 2015) Il Genoma umano Gene codificanti proteine Gene non codificanti proteine Geni codificanti proteine 3 Il
Laboratorio di Bioinformatica I Introduzione alla Genomica Dott. Sergio Marin Vargas (2014 / 2015) Il Genoma umano Gene codificanti proteine Gene non codificanti proteine Geni codificanti proteine 3 Il
METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA
 METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA Scopo di questa esercitazione è apprendere l utilizzo di internet per: STUDIO DELLA STRUTTURA E DELLA FUNZIONE DELLE PROTEINE Conoscere i database
METODOLOGIE BIOCHIMICHE ESERCITAZIONE DI BIOINFORMATICA Scopo di questa esercitazione è apprendere l utilizzo di internet per: STUDIO DELLA STRUTTURA E DELLA FUNZIONE DELLE PROTEINE Conoscere i database
Principali Database biologici
 Principali Database biologici Acidi nucleici: -Sequenze DNA genomico -Sequenze di trascritti (mrna) La maggior quantità di dati biologici presenti nei database è rappresentata da sequenze di acidi nucleici
Principali Database biologici Acidi nucleici: -Sequenze DNA genomico -Sequenze di trascritti (mrna) La maggior quantità di dati biologici presenti nei database è rappresentata da sequenze di acidi nucleici
Modulo Laboratorio A.A. 2014/2015
 Biochimica - Laboratorio di Bioinformatica I (CdL. Bioinformatica) Bioinformatica e banche dati biologiche (CdL. Biotecnologie) Modulo Laboratorio A.A. 2014/2015 Docente: Dr. Sergio Marin Vargas Mail:
Biochimica - Laboratorio di Bioinformatica I (CdL. Bioinformatica) Bioinformatica e banche dati biologiche (CdL. Biotecnologie) Modulo Laboratorio A.A. 2014/2015 Docente: Dr. Sergio Marin Vargas Mail:
Lezione 6. Analisi di sequenze biologiche e ricerche in database
 Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Evoluzione del genoma. Silvia Fuselli, 29 novembre 2011
 Evoluzione del genoma Silvia Fuselli, fss@unife.it 29 novembre 2011 In questa lezione parleremo di Meccanismi di evoluzione del genoma Formazione di nuovi geni Dimensioni del genoma e complessità degli
Evoluzione del genoma Silvia Fuselli, fss@unife.it 29 novembre 2011 In questa lezione parleremo di Meccanismi di evoluzione del genoma Formazione di nuovi geni Dimensioni del genoma e complessità degli
BLAST: Basic Local Alignment Search Tool
 BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
II LEZIONE. Database di interesse per la genetica e la biologia molecolare. Portali per l'accesso a database e servizi bioinformatici
 II LEZIONE Database di interesse per la genetica e la biologia molecolare Portali per l'accesso a database e servizi bioinformatici DATABASE DI GENETICA E BIOLOGIA MOLECOLARE OMIM Online Mendelian Inheritance
II LEZIONE Database di interesse per la genetica e la biologia molecolare Portali per l'accesso a database e servizi bioinformatici DATABASE DI GENETICA E BIOLOGIA MOLECOLARE OMIM Online Mendelian Inheritance
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
50 kb 4-5 milioni milioni 100 milioni 165 milioni Fago E. Coli S. cerevisiae C. elegans D. melanogaster. Human 3 miliardi
 Genomi GENOMI 50 kb 4-5 milioni 12-13 milioni 100 milioni 165 milioni Fago E. Coli S. cerevisiae C. elegans D. melanogaster Human 3 miliardi Problematiche etiche, privacy, scelte lavorative, rapporto
Genomi GENOMI 50 kb 4-5 milioni 12-13 milioni 100 milioni 165 milioni Fago E. Coli S. cerevisiae C. elegans D. melanogaster Human 3 miliardi Problematiche etiche, privacy, scelte lavorative, rapporto
Vai al sito: Incolla nel box vuoto la sequenza nucleotidica
 Identificare il gene a cui appartiene la sequenza (sonda) e la sua posizione sul cromosoma. Per raggiungere l obiettivo della prima parte dell attività devi usare il software BLAT (BLAST- Like Alignment
Identificare il gene a cui appartiene la sequenza (sonda) e la sua posizione sul cromosoma. Per raggiungere l obiettivo della prima parte dell attività devi usare il software BLAT (BLAST- Like Alignment
Bioinformatica (3) Banche dati biologiche. Dott. Alessandro Laganà
 Banche dati biologiche. Dott. Alessandro Laganà") Bioinformatica (3) Banche dati biologiche Dott. Alessandro Laganà Banche dati biologiche Organismi e sequenze biologiche Rappresentazione digitale dei dati biologici e formati Banche dati generiche: NCBI,
Bioinformatica (3) Banche dati biologiche Dott. Alessandro Laganà Banche dati biologiche Organismi e sequenze biologiche Rappresentazione digitale dei dati biologici e formati Banche dati generiche: NCBI,
UTILIZZO DI BLAST PER ALCUNE SEMPLICI APPLICAZIONI IN STUDI GENOMICI
 UTILIZZO DI BLAST PER ALCUNE SEMPLICI APPLICAZIONI IN STUDI GENOMICI Come prima cosa diamo un occhiata alla nostra sequenza di interesse, chiamata «unknown sequence» Con un doppio click possiamo visualizzarla
UTILIZZO DI BLAST PER ALCUNE SEMPLICI APPLICAZIONI IN STUDI GENOMICI Come prima cosa diamo un occhiata alla nostra sequenza di interesse, chiamata «unknown sequence» Con un doppio click possiamo visualizzarla
SRS (Sequence Retrieval System) della EBI che mette a disposizione anche dello spazio sul server per memorizzare le richerche.
 della EBI che mette a disposizione anche dello spazio sul server per memorizzare le richerche.") I due centri maggiori, EBI e NCBI hanno sviluppato sistemi dedicati di RETRIEVAL allo scopo di ottenere il massimo delle informazioni con il minimo sforzo da parte dell utente SRS (Sequence Retrieval System)
I due centri maggiori, EBI e NCBI hanno sviluppato sistemi dedicati di RETRIEVAL allo scopo di ottenere il massimo delle informazioni con il minimo sforzo da parte dell utente SRS (Sequence Retrieval System)
Informatica e biotecnologie I parte
 Informatica e biotecnologie I parte Banche dati biologiche Bioinformatica La Bioinformatica è una disciplina che affronta con metodiche proprie delle Scienze dell'informazione problemi propri della Biologia.
Informatica e biotecnologie I parte Banche dati biologiche Bioinformatica La Bioinformatica è una disciplina che affronta con metodiche proprie delle Scienze dell'informazione problemi propri della Biologia.
MODELLO SCHEDA INSEGNAMENTO. II II Luigi Cerulo
 Corso di L/LM/LMCU Denominazione insegnamento: MODELLO SCHEDA INSEGNAMENTO Numero di Crediti: 6 Anno: Semestre: Docente Titolare: Scienze e Tecnologie Genetiche Bioinformatica II II Luigi Cerulo Dottorandi/assegnisti
Corso di L/LM/LMCU Denominazione insegnamento: MODELLO SCHEDA INSEGNAMENTO Numero di Crediti: 6 Anno: Semestre: Docente Titolare: Scienze e Tecnologie Genetiche Bioinformatica II II Luigi Cerulo Dottorandi/assegnisti
Tecnologia del DNA ricombinante
 Tecnologia del DNA ricombinante Scoperte rivoluzionarie che hanno permesso lo studio del genoma e della funzione dei singoli geni Implicazioni enormi nel progresso della medicina: comprensione malattie
Tecnologia del DNA ricombinante Scoperte rivoluzionarie che hanno permesso lo studio del genoma e della funzione dei singoli geni Implicazioni enormi nel progresso della medicina: comprensione malattie
lezione martedì 6 aprile 2010 aula 2 ore 9:00 corso integrato di Biologia Applicata (BU) ed Ingegneria Genetica (BCM)
 ed Ingegneria Genetica (BCM)") lezione 15-16 martedì 6 aprile 2010 aula 2 ore 9:00 corso integrato di Biologia Applicata (BU) ed Ingegneria Genetica (BCM) R.A.C.E. Con la RT-PCR si amplifica solo un frammento del cdna Se si vuole identificare
lezione 15-16 martedì 6 aprile 2010 aula 2 ore 9:00 corso integrato di Biologia Applicata (BU) ed Ingegneria Genetica (BCM) R.A.C.E. Con la RT-PCR si amplifica solo un frammento del cdna Se si vuole identificare
Struttura dei genomi delle piante
 Struttura dei genomi delle piante Genomi sequenziati Caratteristiche dei genomi delle piante Classi di geni e funzioni Trasposoni e dimensioni dei genomi Sintenia e colinearità Livelli di organizzazione
Struttura dei genomi delle piante Genomi sequenziati Caratteristiche dei genomi delle piante Classi di geni e funzioni Trasposoni e dimensioni dei genomi Sintenia e colinearità Livelli di organizzazione
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Dimensioni dei Genomi Eucariotici
 Dimensioni dei Genomi Eucariotici plasmids viruses bacteria fungi plants algae insects mollusks bony fish amphibians Il Genoma umano è costituito da circa 3 miliardi di bp e contiene un numero di geni
Dimensioni dei Genomi Eucariotici plasmids viruses bacteria fungi plants algae insects mollusks bony fish amphibians Il Genoma umano è costituito da circa 3 miliardi di bp e contiene un numero di geni
ESERCITAZIONE 3. OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST
 ESERCITAZIONE 3 OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST L'esercitazione prevede l'utilizzo di risorse web per effettuare ricerche di similarità con la proteina GRB2 (growth factor
ESERCITAZIONE 3 OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST L'esercitazione prevede l'utilizzo di risorse web per effettuare ricerche di similarità con la proteina GRB2 (growth factor
Principali Database biologici
 Principali Database biologici Acidi nucleici: -Sequenze DNA genomico -Sequenze di trascritti (mrna) La maggior quantità di dati biologici presenti nei database è rappresentata da sequenze di acidi nucleici
Principali Database biologici Acidi nucleici: -Sequenze DNA genomico -Sequenze di trascritti (mrna) La maggior quantità di dati biologici presenti nei database è rappresentata da sequenze di acidi nucleici
Lezione 3. Genoma umano come esempio di genoma eucariote
 Lezione 3 Genoma umano come esempio di genoma eucariote Schema della lezione Sommario degli elementi contenuti in un genoma eucariote Variabilità: dove si trova e come si definisce I grandi progetti internazionali
Lezione 3 Genoma umano come esempio di genoma eucariote Schema della lezione Sommario degli elementi contenuti in un genoma eucariote Variabilità: dove si trova e come si definisce I grandi progetti internazionali
Decode NGS data: search for genetic features
 Decode NGS data: search for genetic features Valeria Michelacci NGS course, June 2015 Blast searches What we are used to: online querying NCBI database for the presence of a sequence of interest ONE SEQUENCE
Decode NGS data: search for genetic features Valeria Michelacci NGS course, June 2015 Blast searches What we are used to: online querying NCBI database for the presence of a sequence of interest ONE SEQUENCE
LA BIOLOGIA MOLECOLARE E UNA BRANCA DELLA BIOLOGIA CHE STUDIA LE BASI MOLECOLARI DELLE FUNZIONI BIOLOGICHE, PONENDO UNA PARTICOLARE ATTENZIONE A QUEI
 CONCETTI DI BASE LA BIOLOGIA MOLECOLARE E UNA BRANCA DELLA BIOLOGIA CHE STUDIA LE BASI MOLECOLARI DELLE FUNZIONI BIOLOGICHE, PONENDO UNA PARTICOLARE ATTENZIONE A QUEI PROCESSI CHE COINVOLGONO GLI ACIDI
CONCETTI DI BASE LA BIOLOGIA MOLECOLARE E UNA BRANCA DELLA BIOLOGIA CHE STUDIA LE BASI MOLECOLARI DELLE FUNZIONI BIOLOGICHE, PONENDO UNA PARTICOLARE ATTENZIONE A QUEI PROCESSI CHE COINVOLGONO GLI ACIDI
Basi di dati biologiche
 Basi di dati biologiche Seminario per il corso di Basi di Dati II Luana Rinaldi luana.rinaldi@gmail.com AGENDA: Introduzione alla bioinformatica; Concetti Biologici; Banche dati biologiche; Collaborazioni
Basi di dati biologiche Seminario per il corso di Basi di Dati II Luana Rinaldi luana.rinaldi@gmail.com AGENDA: Introduzione alla bioinformatica; Concetti Biologici; Banche dati biologiche; Collaborazioni
In molecular terms, a gene commonly is defined as the entire nucleic acid sequence that is necessary for the synthesis of a functional polypeptide.
 In molecular terms, a gene commonly is defined as the entire nucleic acid sequence that is necessary for the synthesis of a functional polypeptide. Lodish et al. Molecular Cell Biology In molecular terms,
In molecular terms, a gene commonly is defined as the entire nucleic acid sequence that is necessary for the synthesis of a functional polypeptide. Lodish et al. Molecular Cell Biology In molecular terms,
DATABASE DI GENETICA E BIOLOGIA MOLECOLARE
 DATABASE DI GENETICA E BIOLOGIA MOLECOLARE OMIM Online Mendelian Inheritance in Man EntrezGene curated sequence and descriptive information about genetic loci GenCards HGMD dbsnp database of human genes,
DATABASE DI GENETICA E BIOLOGIA MOLECOLARE OMIM Online Mendelian Inheritance in Man EntrezGene curated sequence and descriptive information about genetic loci GenCards HGMD dbsnp database of human genes,
Genomi vegetali Da 7x10 7 bp per genoma aploide (130Mbp diploide, 5 cromosomi) di Arabidopsis thaliana alle 1,5x10 11 bp ( Mbp=150Gbp) di una
 di Arabidopsis thaliana alle 1,5x10 11 bp ( Mbp=150Gbp) di una") Genomi vegetali Da 7x10 7 bp per genoma aploide (130Mbp diploide, 5 cromosomi) di Arabidopsis thaliana alle 1,5x10 11 bp (150.000Mbp=150Gbp) di una Liliacea. Tra le graminacee il frumento ha un genoma
Genomi vegetali Da 7x10 7 bp per genoma aploide (130Mbp diploide, 5 cromosomi) di Arabidopsis thaliana alle 1,5x10 11 bp (150.000Mbp=150Gbp) di una Liliacea. Tra le graminacee il frumento ha un genoma
Banche dati biologiche
 Banche dati biologiche Tipi di basi di dati Acidi nucleici GenBank, EMBL Data Library, DNA Data Bank of Japan Sequenze proteiche PIR, Swiss-Prot, TrEMBL, UniProt Strutture Protein Data Bank Pubblicazioni
Banche dati biologiche Tipi di basi di dati Acidi nucleici GenBank, EMBL Data Library, DNA Data Bank of Japan Sequenze proteiche PIR, Swiss-Prot, TrEMBL, UniProt Strutture Protein Data Bank Pubblicazioni
Strategie di annotazione di geni e genomi
 Strategie di annotazione di geni e genomi Dr. Giovanni Emiliani giovanni.emiliani@unifi.it Bioinformatica A.A. 2011-1012 Concetti generali Le nuove tecnologie consentono l ottenimento di una grande mole
Strategie di annotazione di geni e genomi Dr. Giovanni Emiliani giovanni.emiliani@unifi.it Bioinformatica A.A. 2011-1012 Concetti generali Le nuove tecnologie consentono l ottenimento di una grande mole
TRE PAROLE CHIAVE DELLA GENETICA
 TRE PAROLE CHIAVE DELLA GENETICA Questo documento è pubblicato sotto licenza Creative Commons Attribuzione Non commerciale Condividi allo stesso modo http://creativecommons.org/licenses/by-nc-sa/2.5/deed.it
TRE PAROLE CHIAVE DELLA GENETICA Questo documento è pubblicato sotto licenza Creative Commons Attribuzione Non commerciale Condividi allo stesso modo http://creativecommons.org/licenses/by-nc-sa/2.5/deed.it