UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I 1 INTRODUZIONE
|
|
|
- Adriano Pagani
- 6 anni fa
- Visualizzazioni
Transcript


1 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A semestre I 1 INTRODUZIONE
2 Motivazioni dell esistenza della biologia computazionale: Biologia Computazionale è l applicazione di strumenti propri delle scienze dell informazione (es. algoritmi, intelligenza artificiale, databases) a problemi di interesse biologico, biotecnologico e biomedico. Attualmente suscita grande interesse perché: La dimensione dei problemi biologici è sufficiente a motivare lo sviluppo di algoritmi efficienti I problemi sono accessibili (elevata quantità di dati pubblici e letteratura inerente) ed interessanti Le scienze biologiche si avvalgono sempre più spesso di strumenti computazionali
3 Biologia computazionale: Scienze dell Informazione o Biologia? (I) Gli sviluppi delle scienze biomediche, (in particolare per quanto riguarda la biologia molecolare) si verificano ad un ritmo tale da porre seri problemi: La nostra capacità tecnologica di acquisire nuovi dati (spesso in quantità elevate) rende impossibile la loro analisi in assenza di strumenti efficienti.
4 Biologia computazionale: Scienze dell Informazione o Biologia? (II) La biologia computazionale gode di un interesse sempre crescente sia nei dipartimenti di area biomedica che nei dipartimenti di scienze dell informazione. Molti problemi biologici si avvalgono di strumenti informatici (es. assemblaggio di frammenti genomici, analisi di biosequenze, costruzione alberi filogenetici ) Molti strumenti biologici sono stati proposti per rispondere a problemi informatici (avete mai pensato che il DNA offre incredibili possibilità di calcolo parallelo? cercate in google DNA COMPUTING )
5 Attacco al virus SARS La reazione della comunità scientifica alla comparsa del virus SARS in Asia nel Febbraio 2003 illustra il ruolo critico che genomica e informatica giocano nella biologia moderna. Marzo 2003: analisi di dati di microarray di DeRisi rivelano che si tratta di un coronavirus. 12 Aprile 2003: Il Michael Smith Genome Sciences Centre in Canada annuncia il sequenziamento del genoma del virus. 15 Aprile 2003: Microarray SARS-specifici vengono messi in commercio. 1 Maggio 2003: Analisi di predizione delle posizioni dei geni SARS ed analisi del genoma SARS pubblicati in Science. Fine Maggio 2003: Pubblicazione analisi filogenetica per trovare la posizione del virus SARS nella famiglia dei coronavirus. Novembre 2008: Pubblicato esperimento in cui viene riportata la costruzione di un virus SARS sintetico(!)
6 In questa prima parte del corso di Bioinformatica Verranno introdotti PROBLEMI COMPUTAZIONALI inerenti a: Allineamento di sequenze Costruzione di alberi filogenetici Gene finding Accesso diretto a banche dati biologiche e studieremo l APPLICAZIONE degli strumenti disponibili per la soluzione di questi problemi (NBB: l applicazione non è possibile senza il disegno di un esperimento).
7 Natura della Biologia Computazionale (I) Biologia Computazionale è altamente interdisciplinare: questo pone dei problemi. Informatici/Biologi/Biotecnologi/Medici/Fisici/Matematici Esistono vari tipi di scienziati in area biomedica (biologi, biotecnologi, ecologi, medici, ) come in area informatica (algoritmisti, ingegneri del software, ). Questa area di ricerca suscita anche molto interesse da parte di fisici, matematici e statistici. PROBLEMA: NULLA è completamente vero / falso nelle scienze biomediche mentre TUTTO è vero / falso in scienze dell informazione / matematica.
8 Natura della Biologia Computazionale (II) Esistono delle fondamentali differenze culturali tra informatici, matematici e scienziati di area biomedica: Biotecnologi/Medici ecc. : cercano di capire un mondo reale estremamente complicato (e sono consapevoli di averne una visione parziale). Vogliono SCOPRIRE qualcosa Informatici/Matematici ecc. : cercano di costruire dei micromondi organizzati ed ordinati che funzionano da rappresentazioni ridotte del mondo reale. Queste rappresentazioni virtuali del mondo reale sono INDISPENSABILI per sviluppare soluzioni efficienti a problemi altrimenti intrattabili. Vogliono INVENTARE qualcosa
9 Natura della Biologia Computazionale (III) Altre differenze importanti: Scienziati area Biomedica (BM) Scenziati area computazionale o matematica (CM) Conseguenze? Data driven Algorithm driven Pagine web CM migliori di pagine web BM Bagaglio di conoscenze più vasto e più vario (biochimica/genetica/chimic a/fisica ) Sono a loro agio rispetto al fatto che OGNI DATO contiene degli errori Costi sperimentali estremamente elevati (pianificazione sperimentale accurata) Bagaglio di conoscenze più specializzato (sono in grado di svolgere operazioni complesse in modo efficiente) Non lo sono Metriche fondamentali per pianificare un esperimento sono stime del tempo necessario e qualità delle soluzioni prodotte
10 Cercheremo molto spesso di valutare due aspetti degli algoritmi utilizzati in biologia computazionale: Correttezza Efficienza Scienze dell Informazione per scienziati di area biomedica Descriveremo algoritmi che ritornano, in modo DIMOSTRABILE, la miglior soluzione possibile ad un problema combinatorio BEN DEFINITO. Esistono inoltre altre procedure dette EURISTICHE che restituiscono buoni risultati in problemi pratici ma di cui non è dimostrabile la correttezza. Una grossa difficoltà è quella di estrarre problemi ben definiti da problemi reali complessi in modo da rendere questi ultimi affrontabili.
11 Insidie in esperimenti di Biologia Computazionale Spesso (se non sempre) gli esperimenti di biologia computazionale sono svolti in parti separate, ognuna affidata ad una categoria differente di figure professionali. Il rischio maggiore è che le parti svolgano operazioni che non convergano ad una soluzione (o, peggio, che la soluzione finale non risponda ai problemi iniziali). Possibile soluzione: potenziare i contatti tra le parti in gioco in modo che il team Biomedico formalizzi il problema in esame nel modo più stringente possibile ed il team informatico sia quindi in grado di costruire una soluzione adatta al problema e che permetta di salvaguardare contemporaneamente correttezza (quando possibile), efficienza e coerenza della soluzione proposta con il problema in esame.
12 Obiettivi della prima parte del corso Presentazione di problemi di interesse biologico/biotecnologico e della loro formalizzazione. Descrizione di algoritmi di uso corrente utilizzati per rispondere ai problemi presentati (analisi delle caratteristiche dei vari algoritmi). Ricerca sistematica del punto di contatto tra le varie discipline coinvolte nella soluzione dei problemi (questo è l obiettivo fondamentale del corso)

13 (alcune) Sfide della Biologia Computazionale
14 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A semestre I 2 ALLINEAMENTO DI BIOSEQUENZE

15 Elementi conservati nel corso dell evoluzione Kellis et al. Nature 2003 Possiamo LEGGERE l evoluzione per trovare elementi funzionali
16 Obiettivo di oggi: Come possiamo ALLINEARE le sequenze di due geni?
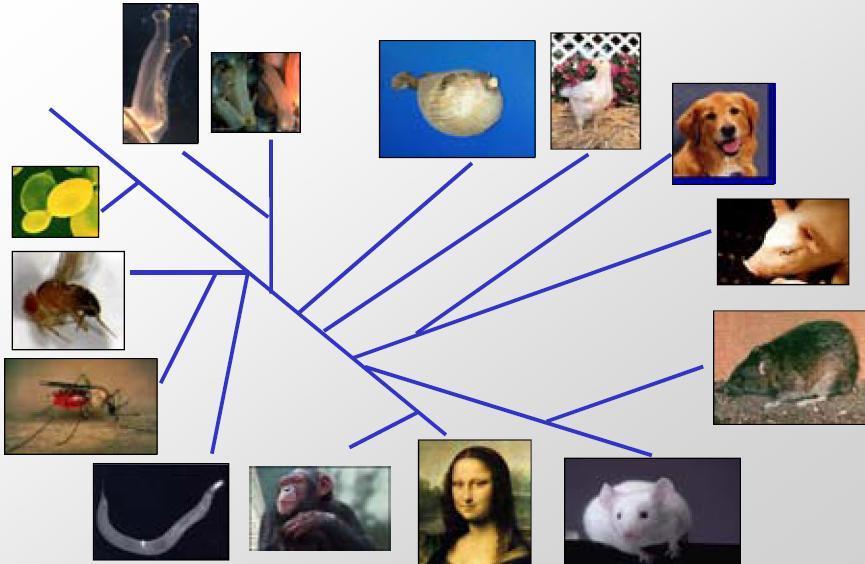
17

18 I genomi cambiano nel tempo Stato iniziale mutazione delezione inserzione Stato finale

19 Obiettivo dell allineamento: Stato iniziale Stato finale INFERENZA DELLE OPERAZIONI DI MODIFICA (editing)
20 Bio CS FORMALIZZAZIONE DEL PROBLEMA 1. Definizione di un set di operazioni evolutive (mutazione, inserzione e delezione) Simmetria delle operazioni (A C, C A ) permette reversibilità rispetto al tempo. Questa è una scelta di disegno della formalizzazione del problema. Eccezione(Bio) : dinucleotidi CpG metilati TpG/CpA (perdita simmetria)
21 Bio CS FORMALIZZAZIONE DEL PROBLEMA 2. Definizione di un criterio di ottimalità (minimo numero operazioni, minimo costo complessivo, ) E IMPOSSIBILE inferire la serie ESATTA di operazioni che portano dalla sequenza A alla sequenza B! Rasoio di Occam: Avendo a disposizione diverse ipotesi equivalentemente competitive rispetto ad un dato problema è buona norma scegliere la più semplice e scartare le altre.
22 Bio CS FORMALIZZAZIONE DEL PROBLEMA 3. Definizione di un algoritmo in grado di raggiungere questa condizione di ottimalità (o in grado di approssimarla) Bio Predicibilità Correttezza Rilevanza Casi speciali Trade-off CS Algoritmi Assunzioni Implementazione Trattabilità Computabilità
? (gap non ammessi)")
23 CS FORMULAZIONE 1 : longest common subsequence (LCS) Date due sequenze S1 ed S2 possibilmente originatesi da una medesima sequenza ancestrale quale è la più lunga sottosequenza in comune (LCS)? (gap non ammessi)
?")
24 CS FORMULAZIONE 2 : longest common subsequence (LCS) Date due sequenze S1 ed S2 possibilmente originatesi da una medesima sequenza ancestrale quale è la più lunga sottosequenza in comune (LCS)? (gap ammessi) E basata su edit distance: Numero di cambiamenti per passare da S1 a S2 Funzione di scoring uniforme
 - Trasversioni (variazioni Pirimidine Purine) - Polimerasi confonde relativamente spesso")
25 CS FORMULAZIONE 3 : Allineamento di sequenze Gap ammessi (penalità fissa): - Operazioni di inserzione e delezione - Medesimo costo per ogni carattere inserito/deleto Penalità variabili per le operazioni di editing: - Transizioni (Pirimidine Pirimidine, Purine Purine) - Trasversioni (variazioni Pirimidine Purine) - Polimerasi confonde relativamente spesso A/G e C/T
26 CS FORMULAZIONE 4,5,6, Molte variazioni: es. Penalità variabili in accordo con il numero di gap (riuscite a proporre delle varianti?)
 - Costo inserzione / delezione - Premio per match Serve algoritmo per provare il miglior allineamento: -")
27 CS Come possiamo costruire il miglior allineamento? Data una funzione di scoring additiva : - Costo mutazioni (AG, CT, altre) - Costo inserzione / delezione - Premio per match Serve algoritmo per provare il miglior allineamento: - Enumerazione di tutti i possibili allineamenti? - Come la realizzereste? - Quanti sono i possibili allineamenti di due sequenze?

28 CS Modi di allineare due sequenze di lunghezza m, n: Per due sequenze di lunghezza n: n Enumerazione Tecnica presentata oggi E E
29 CS PUNTO CRUCIALE: gli score sono additivi: Calcolo RICORSIVO del miglior allineamento: - Per una data coppia di sequenze allineate (S1,S2) il miglior allineamento è:. Miglior allineamento di S1[1..i] e S2[1..j]. + Miglior allineamento di S1[ i..n] e S2[ j..m]

30 CS IDEA : riutilizzo delle operazioni svolte dal calcolatore Sottoproblemi IDENTICI! Possiamo riutilizzare più volte la stessa soluzione.
31 CS SOLUZIONE #1 : Creazione di un DIZIONARIO (le chiavi sono le sequenze) Quando dobbiamo allineare due sequenze cerchiamo una soluzione corrispondente alle due sequenze SE ESISTE: ritorniamo il risultato SE NON ESISTE: - calcoliamo la soluzione - inseriamo la soluzione nel dizionario - restituiamo la soluzione Assicuriamoci di non duplicare i calcoli: è necessario calcolare un sottoallineamento una sola volta LOGICA TOP-DOWN
32 CS SOLUZIONE #2 : PROGRAMMAZIONE DINAMICA Creazione di una grossa tabella con indici i,j - Riempiamola compilando gli score di tutti i possibili sottoallineamenti (calcolati in passi che siano i più piccoli possibili) - ci serviranno tutti i possibili passi dell allineamento (per trovare la soluzione finale) Assicuriamoci di non duplicare i calcoli: è necessario calcolare un sottoallineamento una sola volta Soluzione MOLTO SEMPLICE DAL PUNTO DI VISTA COMPUTAZIONALE! LOGICA BOTTOM-UP
=1, F(n) = F(n-1)+F(n-2) 1,1,2,3,5,8,13,21,34,55,89,144,233,377, Obiettivo: calcolo dell n-esimo numero di")
33 CS Introduzione alla PROGRAMMAZIONE DINAMICA I numeri di Fibonacci sono definiti in maniera ricorsiva: F(0)=1, F(1)=1, F(n) = F(n-1)+F(n-2) 1,1,2,3,5,8,13,21,34,55,89,144,233,377, Obiettivo: calcolo dell n-esimo numero di Fibonacci
34 CS SOLUZIONE #1 : Codice R: fibonacci_td <- function(n){ if(n==1 n==2){ return(1) }else{ return ( fibonacci_td(n-1)+fibonacci_td(n-2) ) } } T(n) = T(n-1)+T(n-2)= ( ) = O(2 n ) complessità esponenziale (in realtà è peggio ancora) LOGICA TOP-DOWN
![CS SOLUZIONE #2 : Codice R: fibonacci_bu <- function(n){ fib_table<-vector() fib_table[1] <- 1 fib_table[2] <- 1 for(i in](/docs-images/72/67821328/images/35-1.jpg "3:(n+1)){ fib_table[i] <- fib_table[i-1]+fib_table[i-2] } } return(fib_table[n]) T(n) = O(n) complessità lineare LOGICA")
35 CS SOLUZIONE #2 : Codice R: fibonacci_bu <- function(n){ fib_table<-vector() fib_table[1] <- 1 fib_table[2] <- 1 for(i in 3:(n+1)){ fib_table[i] <- fib_table[i-1]+fib_table[i-2] } } return(fib_table[n]) T(n) = O(n) complessità lineare LOGICA BOTTOM-UP
 - esprime problemi complessi come insiemi di sottoproblemi più")
36 CS Cosa ci insegna l algoritmo iterativo per il calcolo dell ennesimo numero di Fibonacci? (progr. dinamica) Cosa fa la soluzione iterativa? - rivela sottoproblemi identici - ordina le operazioni in modo da favorirne il riutilizzo - riempie una tabella di risultati (intermedi) - esprime problemi complessi come insiemi di sottoproblemi più semplici L ordine delle operazioni conta - approccio top-down molto lento (risultati intermedi non disponibili) - approccio bottom-up efficace (risolve sottoproblemi, salva risultati, non ricalcola la soluzione ma la costruisce da valori già disponibili)
37 CS Basi della programmazione dinamica: Il problema può essere rappresentato come insieme di sottoproblemi I sottoproblemi non sono sovrapposti (problemi indipendenti che si ripresentano molte volte: calcolo una volta e riutilizzo le soluzioni) In problemi di ottimizzazione reali: - scelte ottimali a livello locale - score sommato rispetto a tutto lo spazio dei sottoproblemi - soluzione: traceback per trovare il percorso migliore tra le soluzioni dei sottoproblemi
38 CS Programmazione dinamica sembra una buona soluzione (finora è stata presentata solo con l esempio dei numeri di Fibonacci) COME POSSIAMO APPLICARLA AL PROBLEMA DELL ALLINEAMENTO DI SEQUENZE?


39 CS PUNTO CRUCIALE: gli score sono additivi: Calcolo RICORSIVO del miglior allineamento: - Per una data coppia di sequenze allineate (S1,S2) il miglior allineamento è:. Miglior allineamento di S1[1..i] e S2[1..j]. + Miglior allineamento di S1[ i..n] e S2[ j..m]
40 CS Programmazione dinamica in pratica: 1. Trovare i parametri della matrice (# dimensioni, variabili) 2. Assicurarsi che lo spazio dei sottoproblemi sia finito - se il riutilizzo non è estensivo può darsi che prog. din. non sia la scelta migliore 3. Ordine di calcolo: i sottorisultati devono essere disponibili quando ci servono - (bottom-up è meglio di top-down ma non è sempre così ovvio) 4. Ricorsione: sovraproblema = F(sottoproblema) 5. Ricordare le scelte effettuate in precedenza ( tipicamente F() include min() o max() ) 6. Iniziamo a calcolare: - Riempiamo la matrice di sottorisultati - Definiamo un punto di partenza e ripercorriamo i passi all indietro fino a trovare la migliore soluzione.
41 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Algoritmo più noto per soluzione del problema: Smith-Waterman (1981) Obiettivo: Date due sequenze relativamente lunghe, determinare la sottosequenza della prima sequenza che realizza il maggior grado di similarità con una sottosequenza della seconda sequenza.
42 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Smith-Waterman (1981) 1) Inizializzazione Data una coppia di sequenze S A =a 1,a 2,,a i,a n e S B =b 1,b 2,,b j,b m costruire una matrice H= H i, j, costituita da n+1 righe e m+1 colonne che viene inizializzata ponendo: H i,0 = H 0,j per 0 i n e 0 j m
43 CS 2) Calcolo Bio Smith-Waterman (1981) ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Per ciascuna coppia di elementi a i e b j appartenenti alle sequenze S A e S B si definisce un punteggio di similarità s. Per il confronto di sequenze nucleotidiche tale punteggio viene generalmente definito come segue: s(a i, b j ) = α se a i = b j ( usualmente > 0 ) s(a i, b j ) = β se a i b j ( usualmente 0 ) W k = γ+δ(k-1) dove γ=costo fisso apertura gap δ=penalità aggiuntiva per estensione gap
44 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Smith-Waterman (1981) 2) Calcolo (continua) I valori della matrice H ij vengono determinati secondo l equazione: H i,j = max(h i-1,j-1 + s(a i, b j ), H i-1,j - pgap, H i,j-1 - pgap, 0) NB: in questo esempio invece di usare W k = γ+δ(k-1) come penalità dinamica per i gap di diversa lunghezza abbiamo utilizzato una penalità costante ( pgap ) indipendente dall estensione dei gap.
45 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Smith-Waterman (1981) 2) Calcolo (continua) I valori della matrice H ij vengono determinati secondo l equazione: H i,j = max(h i-1,j-1 + s(a i, b j ), H i-1,j - pgap, H i,j-1 - pgap, 0) Confronto tra delezione delezione nucleotidi in sequenza B in sequenza A (match/mismatch) di lunghezza 1 di lunghezza 1 score
46 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE Smith-Waterman (1981) 2) Calcolo (continua) I valori della matrice H ij vengono determinati secondo l equazione: H i,j = max(h i-1,j-1 + s(a i, b j ), H i-1,j - pgap, H i,j-1 - pgap, 0) INTERPRETAZIONE: Questa formula ricorsiva considera tutte le possibili terminazioni di segmenti allineati alle cui estremità si trovano a i e b j.


47 CS 2) Calcolo Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE

48 CS Bio DUALITA : miglior allineamento miglior percorso attraverso la matrice! NUOVO OBIETTIVO: Trovare il miglior percorso attraverso la matrice!
49 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE 3) Backtracking -Trovare valore massimo - Ripercorrere la matrice fino a quando non raggiungo l angolo in alto a sinistra o trovo 0 sulla diagonale 3.3

50 CS Bio ALLINEAMENTO LOCALE DI SEQUENZE NUCLEOTIDICHE 3) Backtracking -Trovare valore massimo - Ripercorrere la matrice fino a quando non raggiungo l angolo in alto a sinistra o trovo 0 sulla diagonale 3.3 G C C A U U G. G C C U C G
51 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Algoritmo più noto per soluzione del problema: Needleman-Wunsch (1970) Obiettivo: Date due sequenze A e B, determinare il miglior allineamento (avente quindi il maggior grado si similarità tra le sequenze considerate) che comprenda ogni elemento della sequenza A ed ogni elemento della sequenza B.
52 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Algoritmo più noto per soluzione del problema: Needleman-Wunsch (1970) Costrizioni proprie dell allineamento globale: 1) Lo score similarità calcolato confrontando seq A con Seq B, S(A,B),DEVE essere uguale allo score di similarità che si ottiene confrontando seq B con seq A, S(B,A). 2) Io score di similarità tra due sequenze A e B, S(A,B) DEVE essere minore o uguale alla somma degli score di similarità ottenuti confrontando sia seq A che seq B con una terza sequenza (seq C): S(A,B) S(A,C) + S(B,C)
53 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Algoritmo più noto per soluzione del problema: Needleman-Wunsch (1970) Anche l algoritmo di Needleman-Wunsh, come quello di Smith-Waterman, è basato su tecniche di programmazione dinamica.
54 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) 1) Date due sequenze A e B di lunghezza n ed m, rispettivamente, viene costruita una matrice n per m. Nel caso più semplice i valori delle celle M ij vengono inizializzati come segue: 1 (caratteri identici) 0 (altrimenti)

55 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) 2) Una volta inizializzata, la matrice viene trasformata a partire dalla cella in basso a destra. Trasformazione: Aggiungere allo score della cella corrente il massimo valore della celle appartenenti alla coppia riga-colonna avente l angolo in alto a sinistra posizionato in basso e a destra della cella considerata.
 2)")
56 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) 2) Trasformazione
 2)")
57 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) 2) Trasformazione
 si procede verso verso la cella in basso a destra seguendo il percorso che realizza il punteggio più")
58 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) 3) Traceback Partendo dal valore massimo (8) si procede verso verso la cella in basso a destra seguendo il percorso che realizza il punteggio più alto.
 realizzano il massimo punteggio")
59 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) Entrambi i percorsi (allineamenti) realizzano il massimo punteggio (8)
60 CS Bio ALLINEAMENTO GLOBALE DI SEQUENZE Needleman-Wunsch (1970) NB: Non sono ammessi spostamenti diretti verso destra o verso il basso. La soluzione non è unica ( + percorsi con lo score massimo)
61 CS Bio ALLINEAMENTO DI SEQUENZE Allineamento globale : GOTOH 1982 L algoritmo di Needleman-Wunsch è stata la prima soluzione ampiamente utilizzata per risolvere problemi di allineamento globale. Ma, come soluzione, non era efficiente. Nel 1982 Gotho propose una variante simile a quella proposta in precedenza da Smith e Waterman. Lo schema è molto simile ma ci sono alcune differenze importanti.
62 CS Bio ALLINEAMENTO DI SEQUENZE Allineamento globale : GOTOH 1982 Costruzione matrice m per n Inizializzazione: H(0,0) = 0 H(i,0) = i * pgap H(0,j) = j * pgap Riempimento matrice: Come SW ma senza considerare lo 0 (possibili valori < 0) Inizio traceback : sempre da H(m,n)
63 CS Bio ALLINEAMENTO DI SEQUENZE Allineamento globale vs locale


64 CS Bio ALLINEAMENTO DI SEQUENZE Allineamento globale vs locale

65 CS Bio ALLINEAMENTO DI SEQUENZE Allineamento globale vs locale
66 CS Bio ALLINEAMENTO DI SEQUENZE Riassunto: Il processo di allineamento cerca di quantificare la similarità (numero di variazioni esistenti) tra le sequenze confrontate. I metodi presentati si basano su tecniche di programmazione dinamica (soluzione di problemi grandi una volta che questi ultimi sono stati scomposti in problemi più piccoli) Diversi tipi di allineamento: locale e globale
67 Bio ALLINEAMENTO DI SEQUENZE: CS I genomi cambiano nel tempo. Pattern evolutivi simili possono aiutarci a identificare sequenze funzionalmente simili. Confronto di biosequenze (nt, aa) : quantificare la similarità sulla base delle variazioni necessarie per trasformare una sequenza nell altra (allineamento). Molti modi di allineare due sequenze. E necessaria una soluzione per trovare il miglior allineamento possibile. Soluzione efficiente: suddividere il problema in molti sottoproblemi ripetitivi e calcolare le soluzioni dei sottoproblemi seguendo un ordine che garantisca il possibile riutilizzo delle soluzioni già calcolate programmazione dinamica.
68 Bio ALLINEAMENTO DI SEQUENZE: CS Alla fine del processo di allineamento COSA OTTENIAMO? CS: Allineamento con score migliore ( più allineamenti possono avere il massimo score), calcolato in modo efficiente. Bio: UN ALLINEAMENTO E UNA IPOTESI EVOLUTIVA (avente come obiettivo quello di suggerire l esistenza di relazioni funzionali tra le sequenze confrontate)
 > (conservazione crescente durante l evoluzione in caso di omologia funzionale)")
69 Bio ALLINEAMENTO IPOTESI EVOLUTIVA Se è vero che un allineamento equivale ad un ipotesi evolutiva, cosa giustifica tale ipotesi? (dal punto di vista biologico) > (conservazione crescente durante l evoluzione in caso di omologia funzionale)
70 Bio ALLINEAMENTO IPOTESI EVOLUTIVA La sequenza delle proteine è meno conservata rispetto a struttura secondaria, terziaria e quaternaria nel corso dell evoluzione. Questo ha due effetti: 1) Proteine OMOLOGHE possono avere sequenza molto diverse e quindi produrre allineamenti aventi un punteggio di similarità basso. 2) Se la similarità tra due sequenze proteiche è elevata (in maniera statisticamente significativa) è abbastanza ragionevole ipotizzare che tra di esse esista una relazione di omologia funzionale. OMOLOGIA: carattere qualitativo, SIMILARITA : carattere quantitativo
71 Bio ALLINEAMENTO IPOTESI EVOLUTIVA Per ora abbiamo discusso il problema dell allineamento di biosequenze e abbiamo visto le soluzioni algoritmiche. Ma dobbiamo sempre ricordare che: I metodi presentati (Needleman-Wunsch e Smith-Waterman) permettono di trovare il miglior allineamento in modo efficiente ma non garantiscono che il risultato sia biologicamente sensato! Perché un allineamento risulti informativo è di FONDAMENTALE importanza che gli schemi di SCORING abbiano un senso biologico (questo è particolarmente evidente nel caso di allineamenti di sequenze PROTEICHE).
72 Bio ALLINEAMENTO IPOTESI EVOLUTIVA Ha senso parlare di SCORE FISSI per match e mismatch quando cerchiamo di allineare due sequenze proteiche? NO
73 Bio ALLINEAMENTO IPOTESI EVOLUTIVA Ha senso parlare di SCORE FISSI per match e mismatch quando cerchiamo di allineare due sequenze proteiche? NO COME E POSSIBILE DECIDERE QUALE SCORE ASSEGNARE A DEI PARTICOLARI match/mismatch TRA AMINOACIDI? E un problema biologico (si) E un problema informatico (no) è un problema statistico!
74 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat In questa parte discuteremo di: - Punteggi per i match - Penalità per i mismatch (sia nel caso di allineamenti nucleotidici che aminoacidici) - Origine degli score utilizzati in applicazioni pratiche Matrici di sostituzione: -PAM -BLOSUM

75 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat
76 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat Modello probabilistico di allineamento di biosequenze: Ci concentriamo su allineamenti proteici senza gap Dato un allineamento possiamo considerare due opzioni: C: Deriva da sequenze evolutivamente Correlate N: Deriva da sequenze evolutivamente Non correlate Come è possibile distinguere tra i due casi? C è un collegamento tra questa domanda e le matrici di sostituzione amminoacidiche?
77 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat Sequenze evolutivamente NON correlate: Assumiamo che ogni aa in ogni posizione dell allineamento derivi da un campionamento CASUALE di una distribuzione di aa. q a = probabilità dell aminoacido a La probabilità di un allineamento di n caratteri tra le sequenze x ed y è data da:
78 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat Sequenze evolutivamente Correlate: Assumiamo che ogni coppia di aa allineati derivi da un ancestor comune. P ab = probabilità che l evoluzione abbia originato un aminoacido a nella sequenza x ed un aminoacido b nella sequenza y. La probabilità di un allineamento di n caratteri tra le sequenze x ed y è data da:
79 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat Come decidiamo quale dei modelli (C o N) è più probabile dato un allineamento? Consideriamo la probabilità relativa dei modelli dato l allineamento: Calcolando il logaritmo otteniamo:
80 Bio ALLINEAMENTO DI SEQUENZE: INTERPRETAZIONE PROBABILISTICA Stat Gli elementi della matrice degli score di sostituzione per tutte le coppie di aminoacidi a b si calcolano come segue: Ma come calcoliamo i valori P ab (prob. che a e b si siano originati dal medesimo ancestor nel corso dell evoluzione)? DIPENDE DAL TEMPO DI DIVERGENZA: Divergenza recente P ab 0 se a b Divergenza lontana P ab q a q b
81 Bio MATRICI DI SOSTITUZIONE Stat Idea chiave: Collezioni di sequenze che sappiamo a priori essere evolutivamente correlate possono fornire informazioni riguardanti le sostituzioni biologicamente permesse. Questo pone un problema: Come possiamo costruire collezioni di sequenze evolutivamente correlate che criterio possiamo utilizzare per selezionare sequenze evolutivamente correlate?
82 Bio MATRICI DI SOSTITUZIONE Stat MATRICI PAM (Percent Accepted Mutation Matrices) [Dayhoff,1978] Assunzione: analizzando sequenze correlate filogeneticamente (cioè evolutivamente) si può calcolare la probabilità con cui ogni amminoacido subisce un evento di sostituzione, ovvero una point accepted mutation. Furono calcolate le probabilità di sostituzione di ogni amminoacido con ciascun altro, prevedendo in media una mutazione ogni 100 aminoacidi: questa venne chiamata distanza evolutiva 1 PAM. La matrice PAM 1 non ha applicazioni pratiche ma serve per calcolare le matrici PAM di ordine superiore, ottenute simulando successivi passi evolutivi, cioè applicando le probabilità di sostituzione definite in PAM 1. Tipicamente si usano matrici comprese tra PAM 10 (per sequenze filogeneticamente vicine) e PAM 240 (per sequenze filogeneticamente molto lontane).
83 Bio MATRICI DI SOSTITUZIONE Stat BLOSUM (Blocks Amino Acid Substitution Matrices): [Henikoff & Henikoff, 1992] Utilizzano la banca dati BLOCKS che contiene una collezione di allineamenti multipli (senza gap) di segmenti di proteine suddivisi in famiglie. I blocchi corrispondono a regioni strutturalmente conservate! Ogni blocco di allineamenti contiene sequenze con un numero di aminoacidi identici superiore ad una certa percentuale P, solitamente compresa tra il 30% e il 95%. Da ogni blocco è possibile ricavare la frequenza relativa di sostituzione degli aminoacidi, che può essere utilizzata per calcolare la matrice di punteggi. L indice associato alla matrice BLOSUM indica la percentuale di identità minima all interno del blocco.
84 Bio MATRICI DI SOSTITUZIONE Stat PAM vs BLOSUM BLOSUM 45 BLOSUM 62 BLOSUM 90 > Divergenza < Divergenza PAM 250 PAM 160 PAM 100
85 Bio MATRICI DI SOSTITUZIONE Stat PAM vs BLOSUM: quale delle due scegliere? (I) I due tipi di matrici partono da presupposti diversi: PAM: si assume un modello in cui le sostituzioni aminoacidiche osservate a grandi distanze evolutive derivino esclusivamente dalla somma di tante mutazioni indipendenti. I punteggi risultanti esprimono quanto sia più probabile che l appaiamento di una coppia di aminoacidi sia dovuto a omologia piuttosto che al caso. BLOSUM: non sono basate esplicitamente su un modello evolutivo delle mutazioni dei singoli aa. E in grado di catturare meglio la sostenibilità di una certa mutazione dato un contesto (fornito dai blocchi di sequenze corrispondenti a regioni strutturalmente conservate).
86 Bio MATRICI DI SOSTITUZIONE Stat PAM vs BLOSUM: quale delle due scegliere? (II) Il confronto tra le matrici PAM e BLOSUM, a un livello di sostituzioni paragonabili, indica che i due tipi di matrice sono effettivamente molto simili tra loro, ma non uguali. Le PAM tendono a premiare sostituzioni aminoacidiche che derivano da una singola base, penalizzando le sostituzioni che comportano cambi di codone più complessi. Si ritiene che le matrici BLOSUM siano più adatte delle PAM per effettuare ricerche di similarità di sequenze.

87 Bio BLOSUM 50 Stat Sostituzioni a penalità lieve? Isoleucina e Leucina: score di mutazione reciproci > 0 Triptofano: premio di conservazione estremamente elevato
88 ALLINEAMEMTO PAIRWISE Sequenze biologiche variano durante l evoluzione Perché le ipotesi evolutive generate abbiano senso è necessario assegnare degli score di sostituzione che siano statisticamente frequenti nel corso dell evoluzione Bio BIOLOGIA COMPUTAZIONALE: è possibile generare Ipotesi evolutive Stat CS E possibile calcolare in modo efficiente un allineamento che richieda il minor numero variazioni per passare da una sequenza all altra
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
Le sequenze consenso
 Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
q xi Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p i >0;
 Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Programmazione dinamica
 rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
A W T V A S A V R T S I A Y T V A A A V R T S I A Y T V A A A V L T S I
 COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
Bioinformatics more basic notions
 Bioinformatics more basic notions Alcune slides provengono dal materiale rilasciato da: Dr Sergio Marin Vargas - Verona Prof. Riccardo Percudari - Parma Bioinformatics Bio-inspired Computer science Gli
Bioinformatics more basic notions Alcune slides provengono dal materiale rilasciato da: Dr Sergio Marin Vargas - Verona Prof. Riccardo Percudari - Parma Bioinformatics Bio-inspired Computer science Gli
Allineamenti di sequenze: concetti e algoritmi
 Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre II. 2 Banche dati biologiche
 Docente: Giorgio Valentini UNIVERSITÀ DEGLI STUDI DI MILANO Bioinformatica A.A. 2016-2017 semestre II 2 Banche dati biologiche Banche dati biologiche e bioinformatica: Come mai esistono numerosi strumenti
Docente: Giorgio Valentini UNIVERSITÀ DEGLI STUDI DI MILANO Bioinformatica A.A. 2016-2017 semestre II 2 Banche dati biologiche Banche dati biologiche e bioinformatica: Come mai esistono numerosi strumenti
PROGRAMMAZIONE DINAMICA. Prof. Reho Gabriella Olimpiadi di Informatica
 PROGRAMMAZIONE DINAMICA Quando si usa P.D.? La programmazione dinamica si usa nei casi in cui esista una definizione ricorsiva del problema, ma la trasformazione diretta di tale definizione in un algoritmo
PROGRAMMAZIONE DINAMICA Quando si usa P.D.? La programmazione dinamica si usa nei casi in cui esista una definizione ricorsiva del problema, ma la trasformazione diretta di tale definizione in un algoritmo
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
Bioinformatica. Analisi del genoma
 Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni:
: Allineamento di sequenze (parte 1) Antonella Meloni:") Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Distanza di Edit. Speaker: Antinisca Di Marco Data:
 Distanza di Edit Speaker: Antinisca Di Marco Data: 14-04-2016 Confronto di sequenze Il confronto tra sequenze in biologia computazionale è la base per: misurare la similarità tra le sequenze allineamento
Distanza di Edit Speaker: Antinisca Di Marco Data: 14-04-2016 Confronto di sequenze Il confronto tra sequenze in biologia computazionale è la base per: misurare la similarità tra le sequenze allineamento
Algoritmi e Strutture Dati
 Algoritmi Ricorsivi e Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino A.A. 2006/07 I conigli di Fibonacci Ricerca Binaria L isola dei conigli
Algoritmi Ricorsivi e Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino A.A. 2006/07 I conigli di Fibonacci Ricerca Binaria L isola dei conigli
Sistemi di Elaborazione delle Informazioni
 SCUOLA DI MEDICINA E CHIRURGIA Università degli Studi di Napoli Federico II Corso di Sistemi di Elaborazione delle Informazioni Dott. Francesco Rossi a.a. 2016/2017 1 I linguaggi di programmazione e gli
SCUOLA DI MEDICINA E CHIRURGIA Università degli Studi di Napoli Federico II Corso di Sistemi di Elaborazione delle Informazioni Dott. Francesco Rossi a.a. 2016/2017 1 I linguaggi di programmazione e gli
Allineamento e similarità di sequenze
 Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
la edit distance tra X e Y è la distanza relativa all allineamento (o agli allineamenti) che minimizza tale distanza.
 che minimizza tale distanza.") Algoritmica 14/15 EDIT DISTANCE Il problema della edit distance (distanza di edizione, in una versione in italiano scarsamente usata) è alla base dei problemi di confronto fra sequenze perché il meccanismo
Algoritmica 14/15 EDIT DISTANCE Il problema della edit distance (distanza di edizione, in una versione in italiano scarsamente usata) è alla base dei problemi di confronto fra sequenze perché il meccanismo
Algoritmi greedy. Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione
 Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
SISTEMI LINEARI. x y + 2t = 0 2x + y + z t = 0 x z t = 0 ; S 3 : ; S 5x 2y z = 1 4x 7y = 3
 SISTEMI LINEARI. Esercizi Esercizio. Verificare se (,, ) è soluzione del sistema x y + z = x + y z = 3. Trovare poi tutte le soluzioni del sistema. Esercizio. Scrivere un sistema lineare di 3 equazioni
SISTEMI LINEARI. Esercizi Esercizio. Verificare se (,, ) è soluzione del sistema x y + z = x + y z = 3. Trovare poi tutte le soluzioni del sistema. Esercizio. Scrivere un sistema lineare di 3 equazioni
Programmazione dinamica
 Programmazione dinamica Violetta Lonati Università degli studi di Milano Dipartimento di Informatica Laboratorio di algoritmi e strutture dati Corso di laurea in Informatica Violetta Lonati Programmazione
Programmazione dinamica Violetta Lonati Università degli studi di Milano Dipartimento di Informatica Laboratorio di algoritmi e strutture dati Corso di laurea in Informatica Violetta Lonati Programmazione
Corso di Perfezionamento
 Programmazione Dinamica 1 1 Dipartimento di Matematica e Informatica Università di Camerino 15 febbraio 2009 Tecniche di Programmazione Tecniche di progettazione di algoritmi: 1 Divide et Impera 2 Programmazione
Programmazione Dinamica 1 1 Dipartimento di Matematica e Informatica Università di Camerino 15 febbraio 2009 Tecniche di Programmazione Tecniche di progettazione di algoritmi: 1 Divide et Impera 2 Programmazione
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Note per la Lezione 6 Ugo Vaccaro
 Progettazione di Algoritmi Anno Accademico 2016 2017 Note per la Lezione 6 Ugo Vaccaro Ancora sulla tecnica Programmazione Dinamica Nella lezione scorsa abbiamo appreso che la tecnica Divide-et-Impera,
Progettazione di Algoritmi Anno Accademico 2016 2017 Note per la Lezione 6 Ugo Vaccaro Ancora sulla tecnica Programmazione Dinamica Nella lezione scorsa abbiamo appreso che la tecnica Divide-et-Impera,
Esercizi svolti. delle matrici
 Esercizi svolti. astratti. Si dica se l insieme delle coppie reali (x, y) soddisfacenti alla relazione x + y è un sottospazio vettoriale di R La risposta è sì, perchè l unica coppia reale che soddisfa
Esercizi svolti. astratti. Si dica se l insieme delle coppie reali (x, y) soddisfacenti alla relazione x + y è un sottospazio vettoriale di R La risposta è sì, perchè l unica coppia reale che soddisfa
Programmazione dinamica
 Programmazione dinamica Ilaria Castelli castelli@dii.unisi.it Università degli Studi di Siena Dipartimento di Ingegneria dell Informazione A.A. 29/21 I. Castelli Programmazione dinamica, A.A. 29/21 1/35
Programmazione dinamica Ilaria Castelli castelli@dii.unisi.it Università degli Studi di Siena Dipartimento di Ingegneria dell Informazione A.A. 29/21 I. Castelli Programmazione dinamica, A.A. 29/21 1/35
Algoritmi e strutture di dati 2
 Paola Vocca Lezione 4: Programmazione dinamica 1 Caratteristiche Programmazione dinamica: paradigma basato sullo stesso principio utilizzato per il divide et impera o il problema viene decomposto in sotto-problemi
Paola Vocca Lezione 4: Programmazione dinamica 1 Caratteristiche Programmazione dinamica: paradigma basato sullo stesso principio utilizzato per il divide et impera o il problema viene decomposto in sotto-problemi
Lezione 1. Le molecole di base che costituiscono la vita
 Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Gli obiettivi degli algoritmi di allineamento di sequenze di acidi nucleici o proteine sono molteplici. Possiamo ricordare la ricerca di similarità nelle banche dati, la costruzione
ALLINEAMENTO DI SEQUENZE Gli obiettivi degli algoritmi di allineamento di sequenze di acidi nucleici o proteine sono molteplici. Possiamo ricordare la ricerca di similarità nelle banche dati, la costruzione
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Operazioni tra matrici e n-uple
 CAPITOLO Operazioni tra matrici e n-uple Esercizio.. Date le matrici 0 4 e dati λ = 5, µ =, si calcoli AB, BA, A+B, B A, λa+µb. Esercizio.. Per ognuna delle seguenti coppie di matrici A, B e scalari λ,
CAPITOLO Operazioni tra matrici e n-uple Esercizio.. Date le matrici 0 4 e dati λ = 5, µ =, si calcoli AB, BA, A+B, B A, λa+µb. Esercizio.. Per ognuna delle seguenti coppie di matrici A, B e scalari λ,
Simulazione. D.E.I.S. Università di Bologna DEISNet
 Simulazione D.E.I.S. Università di Bologna DEISNet http://deisnet.deis.unibo.it/ Introduzione Per valutare le prestazioni di un sistema esistono due approcci sostanzialmente differenti Analisi si basa
Simulazione D.E.I.S. Università di Bologna DEISNet http://deisnet.deis.unibo.it/ Introduzione Per valutare le prestazioni di un sistema esistono due approcci sostanzialmente differenti Analisi si basa
1) Hamming bound, coset, codici equivalenti
 Hamming bound, coset, codici equivalenti") Argomenti della Lezione ) Hamming bound, coset, codici equivalenti 2) Esercizi sui codici lineari a blocchi Osservazione () Per effettuare la decodifica a rivelazione di errore si può seguire una delle
Argomenti della Lezione ) Hamming bound, coset, codici equivalenti 2) Esercizi sui codici lineari a blocchi Osservazione () Per effettuare la decodifica a rivelazione di errore si può seguire una delle
Markov Chains and Markov Chain Monte Carlo (MCMC)
") Markov Chains and Markov Chain Monte Carlo (MCMC) Alberto Garfagnini Università degli studi di Padova December 11, 2013 Catene di Markov Discrete dato un valore x t del sistema ad un istante di tempo fissato,
Markov Chains and Markov Chain Monte Carlo (MCMC) Alberto Garfagnini Università degli studi di Padova December 11, 2013 Catene di Markov Discrete dato un valore x t del sistema ad un istante di tempo fissato,
Universita degli Studi di Siena
 Universita degli Studi di Siena Facolta di Ingegneria Dispense del corso di Sistemi di Supporto alle Decisioni I L algoritmo per la risoluzione di problemi di programmazione dinamica Chiara Mocenni Corso
Universita degli Studi di Siena Facolta di Ingegneria Dispense del corso di Sistemi di Supporto alle Decisioni I L algoritmo per la risoluzione di problemi di programmazione dinamica Chiara Mocenni Corso
Sistemi lineari. Lorenzo Pareschi. Dipartimento di Matematica & Facoltá di Architettura Universitá di Ferrara
 Sistemi lineari Lorenzo Pareschi Dipartimento di Matematica & Facoltá di Architettura Universitá di Ferrara http://utenti.unife.it/lorenzo.pareschi/ lorenzo.pareschi@unife.it Lorenzo Pareschi (Univ. Ferrara)
Sistemi lineari Lorenzo Pareschi Dipartimento di Matematica & Facoltá di Architettura Universitá di Ferrara http://utenti.unife.it/lorenzo.pareschi/ lorenzo.pareschi@unife.it Lorenzo Pareschi (Univ. Ferrara)
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
L A B C di R. Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010
 L A B C di R 0 20 40 60 80 100 2 3 4 5 6 7 8 Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010 La scelta del test statistico giusto La scelta della analisi
L A B C di R 0 20 40 60 80 100 2 3 4 5 6 7 8 Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010 La scelta del test statistico giusto La scelta della analisi
Tecniche Algoritmiche: divide et impera
 Tecniche Algoritmiche: divide et impera Una breve presentazione F. Damiani - Alg. & Lab. 04/05 Divide et impera (o Divide and conquer) Per regnare occorre tenere divisi i nemici e trarne vantaggio F. Damiani
Tecniche Algoritmiche: divide et impera Una breve presentazione F. Damiani - Alg. & Lab. 04/05 Divide et impera (o Divide and conquer) Per regnare occorre tenere divisi i nemici e trarne vantaggio F. Damiani
Introduzione al Corso di Algoritmi
 Introduzione al Corso di Algoritmi Di cosa parliamo oggi: Una discussione generale su cosa studieremo, perchè lo studeriemo, come lo studieremo,... Un esempio illustrativo di cosa studeriemo Informazione
Introduzione al Corso di Algoritmi Di cosa parliamo oggi: Una discussione generale su cosa studieremo, perchè lo studeriemo, come lo studieremo,... Un esempio illustrativo di cosa studeriemo Informazione
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
2.6 Calcolo degli equilibri di Nash
 92 2 Giochi non Cooperativi Per queste estensioni di giochi non finiti si possono provare risultati analoghi a quelli visti per i giochi finiti. Rimandiamo alla bibliografia per uno studio più approfondito
92 2 Giochi non Cooperativi Per queste estensioni di giochi non finiti si possono provare risultati analoghi a quelli visti per i giochi finiti. Rimandiamo alla bibliografia per uno studio più approfondito
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
Misure di diversità tra unità statistiche. Loredana Cerbara
 Misure di diversità tra unità statistiche Loredana Cerbara LA DISTANZA IN STATISTICA In statistica la distanza ha un significato diverso da quello che si può intuire in altre discipline, dove, peraltro,
Misure di diversità tra unità statistiche Loredana Cerbara LA DISTANZA IN STATISTICA In statistica la distanza ha un significato diverso da quello che si può intuire in altre discipline, dove, peraltro,
Edit distance. v intner RIMDMDMMI wri t ers
 L'allineamento Edit distance Le operazioni permesse sono: I: insert (inserimento, inserzione) D: delete (cancellazione, delezione, rimozione) R: replacement (substition, sostituzione) M: match (corrispondenza,
L'allineamento Edit distance Le operazioni permesse sono: I: insert (inserimento, inserzione) D: delete (cancellazione, delezione, rimozione) R: replacement (substition, sostituzione) M: match (corrispondenza,
Metodi di Distanza. G.Allegrucci riproduzione vietata
 Metodi di Distanza La misura più semplice della distanza tra due sequenze nucleotidiche è contare il numero di siti nucleotidici che differiscono tra le due sequenze Quando confrontiamo siti omologhi in
Metodi di Distanza La misura più semplice della distanza tra due sequenze nucleotidiche è contare il numero di siti nucleotidici che differiscono tra le due sequenze Quando confrontiamo siti omologhi in
COME CALCOLARE IL PUNTEGGIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi
 COME CALCOLARE IL PUNTEGGIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
COME CALCOLARE IL PUNTEGGIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
Parte III: Algoritmo di Branch-and-Bound
 Parte III: Algoritmo di Branch-and-Bound Divide et Impera Sia z * max {c T x : x S} (1) un problema di ottimizzazione combinatoria difficile da risolvere. Domanda: E possibile decomporre il problema (1)
Parte III: Algoritmo di Branch-and-Bound Divide et Impera Sia z * max {c T x : x S} (1) un problema di ottimizzazione combinatoria difficile da risolvere. Domanda: E possibile decomporre il problema (1)
Z-score. lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random
/ deviazione standard random") Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Qualche informazione di carattere organizzativo
 A.A. 2011/12 Qualche informazione di carattere organizzativo Docenti: Alessandra Bianchi e Paolo Dai Pra Orario di ricevimento (Dai Pra): Martedì ore 15:00 e per appuntamento (saranno possibili cambiamenti
A.A. 2011/12 Qualche informazione di carattere organizzativo Docenti: Alessandra Bianchi e Paolo Dai Pra Orario di ricevimento (Dai Pra): Martedì ore 15:00 e per appuntamento (saranno possibili cambiamenti
Anno 4 Matrice inversa
 Anno 4 Matrice inversa 1 Introduzione In questa lezione parleremo della matrice inversa di una matrice quadrata: definizione metodo per individuarla Al termine della lezione sarai in grado di: descrivere
Anno 4 Matrice inversa 1 Introduzione In questa lezione parleremo della matrice inversa di una matrice quadrata: definizione metodo per individuarla Al termine della lezione sarai in grado di: descrivere
4.5 Metodo del simplesso
 4.5 Metodo del simplesso min z = c T x s.v. Ax = b x PL in forma standard Esamina una sequenza di soluzioni di base ammissibili con valori non crescenti della funzione obiettivo fino a raggiungerne una
4.5 Metodo del simplesso min z = c T x s.v. Ax = b x PL in forma standard Esamina una sequenza di soluzioni di base ammissibili con valori non crescenti della funzione obiettivo fino a raggiungerne una
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
Allineamento multiplo
 Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
2.2 Alberi di supporto di costo ottimo
 . Alberi di supporto di costo ottimo Problemi relativi ad alberi hanno numerose applicazioni: progettazione di reti (comunicazione, teleriscaldamento,...) protocolli reti IP memorizzazione compatta di
. Alberi di supporto di costo ottimo Problemi relativi ad alberi hanno numerose applicazioni: progettazione di reti (comunicazione, teleriscaldamento,...) protocolli reti IP memorizzazione compatta di
1 Indipendenza lineare e scrittura unica
 Geometria Lingotto. LeLing7: Indipendenza lineare, basi e dimensione. Ārgomenti svolti: Indipendenza lineare e scrittura unica. Basi e dimensione. Coordinate. Ēsercizi consigliati: Geoling. Indipendenza
Geometria Lingotto. LeLing7: Indipendenza lineare, basi e dimensione. Ārgomenti svolti: Indipendenza lineare e scrittura unica. Basi e dimensione. Coordinate. Ēsercizi consigliati: Geoling. Indipendenza
Come si sceglie l algoritmo di allineamento? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione?
 Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Sviluppo di programmi
 Sviluppo di programmi Per la costruzione di un programma conviene: 1. condurre un analisi del problema da risolvere 2. elaborare un algoritmo della soluzione rappresentato in un linguaggio adatto alla
Sviluppo di programmi Per la costruzione di un programma conviene: 1. condurre un analisi del problema da risolvere 2. elaborare un algoritmo della soluzione rappresentato in un linguaggio adatto alla
Lezioni di Ricerca Operativa
 Lezioni di Ricerca Operativa Massimo Paolucci Dipartimento di Informatica, Sistemistica e Telematica (DIST) Università di Genova paolucci@dist.unige.it Anno accademico 2000/2001 La Ricerca Operativa (Operation
Lezioni di Ricerca Operativa Massimo Paolucci Dipartimento di Informatica, Sistemistica e Telematica (DIST) Università di Genova paolucci@dist.unige.it Anno accademico 2000/2001 La Ricerca Operativa (Operation
Tempo e spazio di calcolo (continua)
") Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
1 Schemi alle differenze finite per funzioni di una variabile
 Introduzione In questa dispensa vengono forniti alcuni elementi di base per la soluzione di equazioni alle derivate parziali che governano problemi al contorno. A questo scopo si introducono, in forma
Introduzione In questa dispensa vengono forniti alcuni elementi di base per la soluzione di equazioni alle derivate parziali che governano problemi al contorno. A questo scopo si introducono, in forma
Omologia di sequenze: allineamento e ricerca
 Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Algoritmi e loro proprietà. Che cos è un algoritmo? Un esempio di algoritmo
 1 Cos è l informatica? L informatica è la scienza della rappresentazione e dell elaborazione dell informazione Algoritmi e loro proprietà Proprietà formali degli Algoritmi Efficienza rispetto al tempo
1 Cos è l informatica? L informatica è la scienza della rappresentazione e dell elaborazione dell informazione Algoritmi e loro proprietà Proprietà formali degli Algoritmi Efficienza rispetto al tempo
Laboratorio di Algoritmi e Strutture Dati
 Laboratorio di Algoritmi e Strutture Dati Docente: Camillo Fiorentini 8 gennaio 8 Il problema è simile all esercizio 5.6 del libro di testo di algoritmi (Introduzione agli algoritmi e strutture dati, T.
Laboratorio di Algoritmi e Strutture Dati Docente: Camillo Fiorentini 8 gennaio 8 Il problema è simile all esercizio 5.6 del libro di testo di algoritmi (Introduzione agli algoritmi e strutture dati, T.
Sistemi II. Sistemi II. Elisabetta Colombo
 Corso di Approfondimenti di Matematica per Biotecnologie, Anno Accademico 2011-2012, http://users.mat.unimi.it/users/colombo/programmabio.html 1 2 3 con R.C.+ o 1.10 Rango massimo e determinante con R.C.+
Corso di Approfondimenti di Matematica per Biotecnologie, Anno Accademico 2011-2012, http://users.mat.unimi.it/users/colombo/programmabio.html 1 2 3 con R.C.+ o 1.10 Rango massimo e determinante con R.C.+
Algoritmo di Branch & Bound
 Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Algoritmo di Branch & Bound Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria
Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Algoritmo di Branch & Bound Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria
Algoritmi e Strutture Dati
 Algoritmi Golosi (Greedy) Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino un algoritmo goloso correttezza Problema della selezione di attività
Algoritmi Golosi (Greedy) Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino un algoritmo goloso correttezza Problema della selezione di attività
Tempo e spazio di calcolo (continua)
") Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Il processo inferenziale consente di generalizzare, con un certo grado di sicurezza, i risultati ottenuti osservando uno o più campioni
 La statistica inferenziale Il processo inferenziale consente di generalizzare, con un certo grado di sicurezza, i risultati ottenuti osservando uno o più campioni E necessario però anche aggiungere con
La statistica inferenziale Il processo inferenziale consente di generalizzare, con un certo grado di sicurezza, i risultati ottenuti osservando uno o più campioni E necessario però anche aggiungere con
La ricorsione. Sommario. Fulvio CORNO - Matteo SONZA REORDA Dip. Automatica e Informatica Politecnico di Torino
 La ricorsione Fulvio CORNO - Matteo SONZA REORDA Dip. Automatica e Informatica Politecnico di Torino Sommario! Definizione di ricorsione e strategie divide et impera! Semplici algoritmi ricorsivi! Merge
La ricorsione Fulvio CORNO - Matteo SONZA REORDA Dip. Automatica e Informatica Politecnico di Torino Sommario! Definizione di ricorsione e strategie divide et impera! Semplici algoritmi ricorsivi! Merge
GENOMA. Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine CONTENUTO FUNZIONE. Progetti genoma in centinaia di organismi
 GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
3.4 Metodo di Branch and Bound
 3.4 Metodo di Branch and Bound Consideriamo un generico problema di Ottimizzazione Discreta dove X è la regione ammissibile. (P ) z = max{c(x) : x X} Metodologia generale di enumerazione implicita (Land
3.4 Metodo di Branch and Bound Consideriamo un generico problema di Ottimizzazione Discreta dove X è la regione ammissibile. (P ) z = max{c(x) : x X} Metodologia generale di enumerazione implicita (Land
PROBABILITÀ SCHEDA N. 5 SOMMA E DIFFERENZA DI DUE VARIABILI ALEATORIE DISCRETE
 Matematica e statistica: dai dati ai modelli alle scelte www.dima.unige/pls_statistica Responsabili scientifici M.P. Rogantin e E. Sasso (Dipartimento di Matematica Università di Genova) PROBABILITÀ SCHEDA
Matematica e statistica: dai dati ai modelli alle scelte www.dima.unige/pls_statistica Responsabili scientifici M.P. Rogantin e E. Sasso (Dipartimento di Matematica Università di Genova) PROBABILITÀ SCHEDA
A m n B n p = P m p. 0 1 a b c d. a b. 0 a 0 c Il risultato e lo stesso solo nel caso in cui c = 0 e a = d.
 Matematica II, 220404 Il prodotto di matrici e un operazione parziale che prende in entrata una matrice A ed una matrice B, tali che il numero delle colonne di A sia uguale al numero delle righe di B,
Matematica II, 220404 Il prodotto di matrici e un operazione parziale che prende in entrata una matrice A ed una matrice B, tali che il numero delle colonne di A sia uguale al numero delle righe di B,
Problemi di localizzazione di servizi (Facility Location Problems)
") 9. Problemi di Localizzazione di Servizi 1 Problemi di localizzazione di servizi (Facility Location Problems) Dato un insieme di clienti richiedenti una data domanda di merce e dato un insieme di possibili
9. Problemi di Localizzazione di Servizi 1 Problemi di localizzazione di servizi (Facility Location Problems) Dato un insieme di clienti richiedenti una data domanda di merce e dato un insieme di possibili
Esercizi sui sistemi di equazioni lineari.
 Esercizi sui sistemi di equazioni lineari Risolvere il sistema di equazioni lineari x y + z 6 x + y z x y z Si tratta di un sistema di tre equazioni lineari nelle tre incognite x, y e z Poichè m n, la
Esercizi sui sistemi di equazioni lineari Risolvere il sistema di equazioni lineari x y + z 6 x + y z x y z Si tratta di un sistema di tre equazioni lineari nelle tre incognite x, y e z Poichè m n, la
Fondamenti di Informatica 6. Algoritmi e pseudocodifica
 Vettori e matrici #1 Fondamenti di Informatica 6. Algoritmi e pseudocodifica Corso di Laurea in Ingegneria Civile A.A. 2010-2011 1 Semestre Prof. Giovanni Pascoschi Le variabili definite come coppie
Vettori e matrici #1 Fondamenti di Informatica 6. Algoritmi e pseudocodifica Corso di Laurea in Ingegneria Civile A.A. 2010-2011 1 Semestre Prof. Giovanni Pascoschi Le variabili definite come coppie
Flusso a Costo Minimo
 Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Flusso a Costo Minimo Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria Dal
Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Flusso a Costo Minimo Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria Dal
Cosa è l Informatica?
 Cosa è l Informatica? Scienza degli elaboratori elettronici (Computer Science) Scienza dell informazione Scienza della rappresentazione, memorizzazione, elaborazione e trasmissione dell informazione Elaboratore
Cosa è l Informatica? Scienza degli elaboratori elettronici (Computer Science) Scienza dell informazione Scienza della rappresentazione, memorizzazione, elaborazione e trasmissione dell informazione Elaboratore
Algoritmi e Strutture Dati (Mod. B) Algoritmi Greedy (parte I)
 Algoritmi Greedy (parte I)") Algoritmi e Strutture Dati (Mod. B) Algoritmi Greedy (parte I) Algoritmi greedy Gli algoritmi per problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione
Algoritmi e Strutture Dati (Mod. B) Algoritmi Greedy (parte I) Algoritmi greedy Gli algoritmi per problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione
Corso di Laurea in Fisica. Geometria. a.a Canale 3 Prof. P. Piazza Magiche notazioni
 Corso di Laurea in Fisica. Geometria. a.a. 23-4. Canale 3 Prof. P. Piazza Magiche notazioni Siano V e W due spazi vettoriali e sia T : V W un applicazione lineare. Fissiamo una base B per V ed una base
Corso di Laurea in Fisica. Geometria. a.a. 23-4. Canale 3 Prof. P. Piazza Magiche notazioni Siano V e W due spazi vettoriali e sia T : V W un applicazione lineare. Fissiamo una base B per V ed una base
Differenze divise. Polinomio di interpolazione nella forma di Newton. Proprietà. Se n=0. Simmetria. Ricorsività. Abbiamo un solo punto
 Differenze divise Polinomio di interpolazione nella forma di Newton Se n=0 Abbiamo un solo punto Se n = 1 Abbiamo 2 punti Il polinomio cercato è la retta passante per i due punti Proprietà Simmetria Differenza
Differenze divise Polinomio di interpolazione nella forma di Newton Se n=0 Abbiamo un solo punto Se n = 1 Abbiamo 2 punti Il polinomio cercato è la retta passante per i due punti Proprietà Simmetria Differenza
Esercitazione. 24 Aprile 2012
 Esercitazione 24 Aprile 2012 Il modello di regressione logistica viene utilizzato quando si è interessati a studiare o analizzare la relazione causale tra una variabile dipendente dicotomica e una o più
Esercitazione 24 Aprile 2012 Il modello di regressione logistica viene utilizzato quando si è interessati a studiare o analizzare la relazione causale tra una variabile dipendente dicotomica e una o più
FORMAZIONE DEL LEGAME PEPTIDICO
 AMINOACIDI FORMAZIONE DEL LEGAME PEPTIDICO SEQUENZA AMINOACIDICA DELL INSULINA STRUTTURA SECONDARIA DELLE PROTEINE STRUTTURA TERZIARIA DELLE PROTEINE STRUTTURA QUATERNARIA DELLE PROTEINE Definizione Processi
AMINOACIDI FORMAZIONE DEL LEGAME PEPTIDICO SEQUENZA AMINOACIDICA DELL INSULINA STRUTTURA SECONDARIA DELLE PROTEINE STRUTTURA TERZIARIA DELLE PROTEINE STRUTTURA QUATERNARIA DELLE PROTEINE Definizione Processi
Intelligenza Artificiale
 Intelligenza Artificiale 17 Marzo 2005 Nome e Cognome: Matricola: ESERCIZIO N 1 Ricerca Cieca 5 punti 1.A) Elencare in modo ordinato i nodi (dell'albero sotto) che vengono scelti per l'espansione dalle
Intelligenza Artificiale 17 Marzo 2005 Nome e Cognome: Matricola: ESERCIZIO N 1 Ricerca Cieca 5 punti 1.A) Elencare in modo ordinato i nodi (dell'albero sotto) che vengono scelti per l'espansione dalle
4.1 Localizzazione e pianificazione delle base station per le reti UMTS
 esercitazione Ottimizzazione Prof E Amaldi Localizzazione e pianificazione delle base station per le reti UMTS Consideriamo il problema di localizzare un insieme di stazioni radio base, base station (BS),
esercitazione Ottimizzazione Prof E Amaldi Localizzazione e pianificazione delle base station per le reti UMTS Consideriamo il problema di localizzare un insieme di stazioni radio base, base station (BS),
Riconoscimento automatico di oggetti (Pattern Recognition)
") Riconoscimento automatico di oggetti (Pattern Recognition) Scopo: definire un sistema per riconoscere automaticamente un oggetto data la descrizione di un oggetto che può appartenere ad una tra N classi
Riconoscimento automatico di oggetti (Pattern Recognition) Scopo: definire un sistema per riconoscere automaticamente un oggetto data la descrizione di un oggetto che può appartenere ad una tra N classi
Lezione 6. Analisi di sequenze biologiche e ricerche in database
 Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
3.6 Metodi basati sui piani di taglio
 3.6 Metodi basati sui piani di taglio Problema generale di Programmazione Lineare Intera (PLI) con A matrice m n e b vettore n 1 razionali min{ c t x : x X = {x Z n + : Ax b} } Sappiamo che esiste una
3.6 Metodi basati sui piani di taglio Problema generale di Programmazione Lineare Intera (PLI) con A matrice m n e b vettore n 1 razionali min{ c t x : x X = {x Z n + : Ax b} } Sappiamo che esiste una
Problemi, algoritmi, calcolatore
 Problemi, algoritmi, calcolatore Informatica e Programmazione Ingegneria Meccanica e dei Materiali Università degli Studi di Brescia Prof. Massimiliano Giacomin Problemi, algoritmi, calcolatori Introduzione
Problemi, algoritmi, calcolatore Informatica e Programmazione Ingegneria Meccanica e dei Materiali Università degli Studi di Brescia Prof. Massimiliano Giacomin Problemi, algoritmi, calcolatori Introduzione
Lezione 6 Richiami di Geometria Analitica
 1 Piano cartesiano Lezione 6 Richiami di Geometria Analitica Consideriamo nel piano due rette perpendicolari che si intersecano in un punto O Consideriamo ciascuna di queste rette come retta orientata
1 Piano cartesiano Lezione 6 Richiami di Geometria Analitica Consideriamo nel piano due rette perpendicolari che si intersecano in un punto O Consideriamo ciascuna di queste rette come retta orientata
Complementi ed Esercizi di Informatica Teorica II
 Complementi ed Esercizi di Informatica Teorica II Vincenzo Bonifaci 21 maggio 2008 4 Problemi di ottimizzazione: il Bin Packing Il problema bin packing è il seguente: dato un insieme di n oggetti di dimensioni
Complementi ed Esercizi di Informatica Teorica II Vincenzo Bonifaci 21 maggio 2008 4 Problemi di ottimizzazione: il Bin Packing Il problema bin packing è il seguente: dato un insieme di n oggetti di dimensioni
Corso di Laurea Specialistica in Ingegneria Informatica
 UNIVERSITÀ DEGLI STUDI DI PADOVA FACOLTÀ DI INGEGNERIA Corso di Laurea Specialistica in Ingegneria Informatica Ragionamento Qualitativo e Apprendimento Automatico per l'analisi di Dati di Genomica RELATORE:
UNIVERSITÀ DEGLI STUDI DI PADOVA FACOLTÀ DI INGEGNERIA Corso di Laurea Specialistica in Ingegneria Informatica Ragionamento Qualitativo e Apprendimento Automatico per l'analisi di Dati di Genomica RELATORE:
Esercizi vari. Alberto Montresor. 19 Agosto, 2014
 Esercizi vari Alberto Montresor 19 Agosto, 2014 Alcuni degli esercizi che seguono sono associati alle rispettive soluzioni. Se il vostro lettore PDF lo consente, è possibile saltare alle rispettive soluzioni
Esercizi vari Alberto Montresor 19 Agosto, 2014 Alcuni degli esercizi che seguono sono associati alle rispettive soluzioni. Se il vostro lettore PDF lo consente, è possibile saltare alle rispettive soluzioni
TEORIA DEI SISTEMI SISTEMI LINEARI
 TEORIA DEI SISTEMI Laurea Specialistica in Ingegneria Meccatronica Laurea Specialistica in Ingegneria Gestionale Indirizzo Gestione Industriale TEORIA DEI SISTEMI SISTEMI LINEARI Ing. Cristian Secchi Tel.
TEORIA DEI SISTEMI Laurea Specialistica in Ingegneria Meccatronica Laurea Specialistica in Ingegneria Gestionale Indirizzo Gestione Industriale TEORIA DEI SISTEMI SISTEMI LINEARI Ing. Cristian Secchi Tel.
Algoritmo dibranch & Bound
 Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Algoritmo dibranch & Bound Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria
Sapienza Università di Roma - Dipartimento di Ingegneria Informatica, Automatica e Gestionale Algoritmo dibranch & Bound Docente: Renato Bruni bruni@dis.uniroma.it Corso di: Ottimizzazione Combinatoria
MATEMATICA PER LO STUDIO DELLE INTERAZIONI STRATEGICHE: TEORIA DEI GIOCHI. Anna TORRE
 MATEMATICA PER LO STUDIO DELLE INTERAZIONI STRATEGICHE: TEORIA DEI GIOCHI Anna TORRE Dipartimento di Matematica, Università di Pavia, Via Ferrata 1, 27100, Pavia, Italy. E-mail: anna.torre@unipv.it 1 SOLUZIONI:
MATEMATICA PER LO STUDIO DELLE INTERAZIONI STRATEGICHE: TEORIA DEI GIOCHI Anna TORRE Dipartimento di Matematica, Università di Pavia, Via Ferrata 1, 27100, Pavia, Italy. E-mail: anna.torre@unipv.it 1 SOLUZIONI: