AIFA POSITION PAPER I FARMACI BIOSIMILARI
|
|
|
- Giuditta Pagano
- 5 anni fa
- Visualizzazioni
Transcript
1 AIFA POSITION PAPER I FARMACI BIOSIMILARI Preparazione del Draft Presentazione del Draft e rilascio per la consultazione pubblica Inizio della consultazione pubblica Fine della consultazione pubblica Versione definitiva
2 Premessa In considerazione dell importanza che i medicinali biologici, inclusi i biosimilari, rivestono per il trattamento di numerose patologie gravi e potenzialmente letali, per molte delle quali in passato non era disponibile alcuna opzione terapeutica efficace, l Agenzia Italiana del Farmaco (AIFA) ritiene indispensabile fornire un documento il cui obiettivo è quello di promuovere la conoscenza e l utilizzo dei biosimilari fornendo, agli operatori sanitari e ai cittadini, informazioni autorevoli, chiare, trasparenti, convalidate ed obiettive, relativamente ai seguenti aspetti: Definizione e principali criteri di caratterizzazione dei medicinali biologici e biosimilari; Inquadramento delle normative regolatorie vigenti in EU in merito ai medicinali biosimilari; Ruolo dei biosimilari nella sostenibilità economica del servizio sanitario nazionale (SSN). Introduzione Le tecniche biotecnologiche hanno permesso lo sviluppo di trattamenti per un ampia varietà di malattie di grande rilievo clinico ed epidemiologico fornendo risposte fondamentali alla crescente domanda di salute della popolazione. Ad oggi, a livello mondiale, milioni di pazienti hanno già beneficiato dei medicinali biologici approvati per la cura o la prevenzione di molte malattie quali, ad esempio, tumori, malattie infiammatorie, autoimmuni, neurologiche e degenerative. Le terapie derivanti dalle biotecnologie sono gravate da enormi sforzi ed investimenti in tutti gli stadi che ne caratterizzano lo sviluppo, dalle fasi di ricerca al sofisticato processo produttivo, fino all accesso al mercato e alla dispensazione delle cure. Ciò pone un problema rilevante in termini di sostenibilità economica per identificare e definire la migliore allocazione delle risorse disponibili. Se da un lato un vasto numero di farmaci biotecnologici è in fase di sviluppo clinico, dall altro la prima generazione di farmaci biologici ha già superato o è in procinto di superare, la scadenza brevettuale. La perdita della copertura brevettuale permette l entrata sulla scena terapeutica dei farmaci cosiddetti biosimilari, medicinali simili ai farmaci biologici originatori non più soggetti a copertura brevettuale, che possono essere prodotti dalle industrie farmaceutiche secondo procedure e normative espresse da specifiche linee guida europee e commercializzati a prezzi inferiori rispetto ai prodotti originatori. I farmaci biosimilari sono, quindi, medicinali biologici autorizzati dall Agenzia Europea dei Medicinali (European Medicines Agency EMA) simili per qualità, efficacia e sicurezza al prodotto biologico di riferimento. La disponibilità dei prodotti Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 2
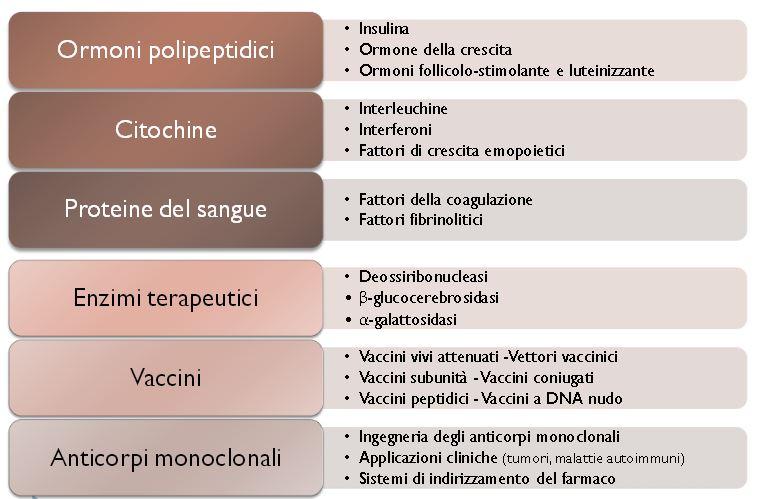
3 biosimilari rappresenta un fattore importante per il mantenimento della sostenibilità economica dei servizi sanitari nel prossimo futuro. I medicinali di origine biologica Secondo la definizione di medicinale biologico prodotta dall EMA Un medicinale biologico è quello che contiene una o più sostanze attive derivate da una fonte biologica; alcune di queste sostanze attive possono essere già presenti nell organismo umano ad esempio proteine come l insulina, l ormone della crescita e l eritropoietina. I medicinali biologici sono molecole più grandi e più complesse rispetto ai medicinali non biologici. Soltanto gli organismi viventi sono in grado di riprodurre tale complessità (EMA/837505/2011). I medicinali biologici, intendendo con tale termine anche quelli biotecnologici, cioè ottenuti con biotecnologie, sono, quindi, farmaci il cui principio attivo è rappresentato da una sostanza prodotta o estratta da un sistema biologico tali prodotti sono a volte definiti medicinali biologici in senso stretto oppure derivata da una sorgente biologica attraverso procedimenti di biotecnologia, comprendenti le tecnologie di DNA ricombinante, l espressione controllata di geni codificanti proteine biologicamente attive nei procarioti o negli eucarioti, metodi a base di ibridomi e di anticorpi monoclonali o biotecnologici (Doc. Ref. EMEA/74562/2006 Rev1). Appartengono alla categoria dei farmaci biologici prodotti quali ormoni ed enzimi, emoderivati e medicinali immunologici come sieri e vaccini, immunoglobuline ed allergeni, oppure anticorpi monoclonali. I medicinali sintetizzati per via biotecnologica differiscono dalle sostanze attive sintetizzate tramite metodiche di chimica farmaceutica tradizionale per molti aspetti, tra i quali, ad esempio, la dimensione molecolare, la complessità strutturale, la stabilità del prodotto finale e la possibilità di differenti modifiche co- e post-traduzionali rilevanti (ad esempio, del profilo di glicosilazione). Inoltre, mentre i farmaci tradizionali costituiti da piccole molecole sono prodotti tramite sintesi chimica, la maggior parte dei farmaci biologici, essendo prodotti tramite biotecnologie che operano su sistemi viventi (microrganismi o cellule animali), presentano numerosi aspetti di eterogeneità legati alla cellula ospite utilizzata, ai plasmidi impiegati per transfettare la cellula ospite e, quindi, trasferire il gene necessario al fine di indurre l espressione della proteina voluta, nonché alle condizioni di crescita e fermentazione e alle differenti metodiche di purificazione. Tutti questi materiali e procedure introducono elementi di differenziazione e non sono Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 3
4 immediatamente trasferibili da un laboratorio ad un altro, contribuendo a determinare l unicità del prodotto. Il processo di produzione di tali farmaci è talmente caratterizzante che si può affermare che il prodotto è il processo di produzione (Karson KL., Nature Biotecnol, 2005). La struttura molecolare e il processo di produzione dei biologici giocano un ruolo importante nel determinare il potenziale immunogenico di questi medicinali, infatti, un ulteriore caratteristica fondamentale dei prodotti biologici è la loro immunogenicità, definita come la capacità di indurre una reazione immunitaria nell organismo: tali molecole hanno la potenzialità di essere riconosciute come non-self dall organismo del paziente ed essere, in qualche caso, neutralizzate nel loro effetto, riducendo l efficacia della terapia. Nel caso, ad esempio, dei vaccini l immunogenicità costituisce, al contrario, la base della strategia terapeutica, inducendo una risposta immunitaria volta a riconoscere e combattere la sostanza contro cui il vaccino è diretto. La maggior parte delle risposte immunitarie indesiderate indotte dall immunogenicità dei prodotti biologici sono moderate e non producono effetti negativi nel paziente. In rari casi, tuttavia, possono verificarsi risposte immunitarie che conducono ad effetti nocivi gravi a detrimento della salute e della sicurezza del paziente. Da quanto esposto è evidente come i farmaci biologici, per la variabilità intrinseca delle molecole e per la complessità delle tecniche di produzione, siano particolarmente difficili da caratterizzare e da riprodurre, al punto tale che alcune differenze possono sussistere anche tra i diversi lotti di uno stesso prodotto, soprattutto se si sono verificate variazioni nelle condizioni di produzione. Per questo motivo le Autorità regolatorie richiedono per la caratterizzazione e il controllo di qualità e di sicurezza dei medicinali biologici, oltre agli esami fisico-chimico-biologici, anche informazioni specifiche sul processo di produzione e su potenziale immunogenico e problemi di sicurezza che da questo possono derivare e controllano in maniera rigorosa che ogni processo della filiera di produzione e di distribuzione di questi farmaci avvenga in ottemperanza alle specifiche linee guida. I Medicinali Biosimilari Con il termine biosimilare viene indicato un medicinale simile a un prodotto biologico di riferimento già autorizzato nell Unione Europea e per il quale sia scaduta la copertura brevettuale. Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 4
5 In particolare, il concetto di medicinale biologico simile è stato introdotto nella legislazione dell'unione Europea dalla direttiva 2001/83/CE e successive modificazioni 1, che all articolo 10(4) ha fornito una definizione implicita di prodotto biosimilare, successivamente recepita nella normativa italiana tramite il decreto legislativo n.219/2006 all articolo 10 punto 7, come segue: Quando un medicinale biologico simile a un medicinale biologico di riferimento non soddisfa le condizioni della definizione di medicinale generico a causa, in particolare, di differenze attinenti alle materie prime o di differenze nei processi di produzione del medicinale biologico e del medicinale biologico di riferimento, il richiedente è tenuto a fornire i risultati delle appropriate prove precliniche o delle sperimentazioni cliniche relative a dette condizioni. I dati supplementari da fornire soddisfano i criteri pertinenti di cui all'allegato tecnico sulla domanda di AIC e le relative linee guida. Non è necessario fornire i risultati delle altre prove e sperimentazioni contenuti nel dossier del medicinale di riferimento. Se i risultati presentati non sono ritenuti sufficienti a garantire l'equivalenza del biogenerico o biosimilare con il medicinale biologico di riferimento è presentata una domanda nel rispetto di tutti i requisiti previsti dall'articolo Nel settembre 2012 l EMA ha diffuso un documento in cui è fornita la seguente definizione: Per medicinale biosimilare si intende un medicinale sviluppato in modo da risultare simile a un medicinale biologico che è già stato autorizzato (il così detto medicinale di riferimento ). I medicinali biosimilari, dunque, differiscono dai farmaci generici che hanno strutture chimiche più semplici e che sono considerati identici ai loro medicinali di riferimento. Il principio attivo di un biosimilare e quello del suo medicinale di riferimento sono di fatto la stessa sostanza biologica, tuttavia possano essere presenti differenze minori dovute alla loro natura complessa e alle tecniche di produzione. Come il medicinale di riferimento, il biosimilare presenta un certo grado di variabilità naturale. Un biosimilare viene approvato quando è stato dimostrato che tale variabilità naturale ed eventuali differenze rispetto al medicinale di riferimento non influiscono sulla sicurezza o sull efficacia. (Questions and Answers on biosimilar medicines EMA/837805/2011 del 27 settembre 2012). Un biosimilare e il suo prodotto di riferimento, essendo ottenuti mediante modalità differenti, non sono identici, ma essenzialmente simili in termini di qualità, sicurezza ed efficacia. Va, infine, menzionata la possibilità che, in taluni casi, il farmaco biosimilare possa essere derivato da processi produttivi più innovativi rispetto a quelli del farmaco di riferimento, tali da far sì che il 1 Direttiva 2003/63/CE e Direttiva 2004/27/CE) 2 EMA/940451/2001 EMA Procedural advice for users of the Centralised Procedure for Similar Biological Medicinal Products applications 5 Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari
6 prodotto biosimilare presenti profili di qualità, per esempio in termini di impurezze, persino migliori rispetto all originatore. Normativa vigente in EU in merito ai medicinali biosimilari La normativa europea costituisce il primo esempio di regolamentazione del processo di autorizzazione dei biosimilari. A questa si sono ispirati l Organizzazione mondiale della sanità (OMS) e molti paesi in tutto il mondo, inclusi USA, Canada, Australia e Giappone. Nell Unione Europea, tutte le domande di autorizzazione all'immissione in commercio di medicinali biologici, incluse quelle per i biosimilari, sono esaminate dall EMA attraverso la procedura centralizzata; di conseguenza l'autorizzazione all'immissione in commercio che ne deriva è valida in tutti gli Stati membri dell'unione Europea. Affinché tale procedura possa essere intrapresa, è anzitutto necessario che il prodotto di riferimento, al quale la domanda di autorizzazione all'immissione in commercio di un prodotto biosimilare si riferisce, sia un medicinale che abbia ottenuto un'autorizzazione all'immissione in commercio nell'unione Europea sulla base di un dossier di registrazione completo, in accordo con quanto previsto dall articolo 8 della Direttiva 2001/83/EC. Come previsto dalla normativa e al fine di fornire orientamenti alle industrie produttrici, l'ema ha pubblicato dei Concept Paper e delle linee guida, sia generali per i prodotti biosimilari, sia modulo CTD (Common Technical Document)-specifiche (riguardanti aspetti specifici della dimostrazione della biosimilarità rispetto a parametri di qualità, studi non clinici e clinici), sia specifiche per le singole categorie di medicinali biosimilari (es. eritropoietine, ormone della crescita, G-CSF, anticorpi monoclonali, etc. ). Tali linee guida sono revisionate con cadenza regolare in modo da riflettere l'esperienza acquisita con le procedure di autorizzazione di prodotti biosimilari già registrati, o comunque avviate, e tenere conto dell'evoluzione della scienza e della tecnologia. Le diverse linee guida sono consultabili su una pagina dedicata del sito EMA al seguente link: 08.jsp&mid=WC0b01ac c&jsenabled=true Poiché il principio attivo di un medicinale biosimilare è essenzialmente simile ma non identico a quello del medicinale biologico di riferimento, l iter registrativo di un prodotto biosimilare è molto differente da quello dei farmaci equivalenti di medicinali non biologici, per i quali è generalmente sufficiente presentare i risultati degli studi di bioequivalenza (Dir. 2003/63/EC, Ann I, P.II). Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 6
7 La normativa richiede che il programma di ricerca e sviluppo sia volto a dimostrare la biosimilarità intesa come la comparabilità tra un biosimilare ed il suo prodotto di riferimento, attraverso l esercizio di comparabilità, ovvero l insieme di una serie di procedure di confronto graduale (stepwise) che inizia con gli studi di qualità (comparabilità fisico-chimiche e biologiche), e prosegue con la valutazione della comparabilità non-clinica (studi non clinici comparativi) e clinica (studi clinici comparativi) per la valutazione dell efficacia e della sicurezza, includendo lo studio dell immunogenicità. L obiettivo primario dell esercizio di comparabilità è la dimostrazione della similarità ( similarity throughout), attraverso studi disegnati in modo tale da individuare le eventuali differenze di qualità tra il biosimilare e il prodotto di riferimento, e assicurare che queste non si traducano in differenze cliniche rilevanti, in termini di sicurezza ed efficacia, tra i due prodotti. Negli studi di qualità la comparabilità è stabilita con riferimento alla struttura molecolare e deve essere dimostrata tramite una completa caratterizzazione analitica, studi di legame al recettore (se applicabili), biotest e adeguati studi su animali, tutti da eseguire in modo rigorosamente comparativo tra biosimilare e prodotto di riferimento. L esercizio di comparabilità pre-clinica e clinica è effettuato mediante specifici studi controllati di valutazione delle proprietà tossicologiche, del profilo farmacocinetico e farmacodinamico, della sicurezza e dell efficacia clinica. L'esercizio di comparabilità è quindi basato su un robusto confronto testa a testa tra il biosimilare e il medicinale di riferimento secondo specifici standard di qualità, sicurezza ed efficacia, avendo definito a priori le differenze ritenute accettabili in quanto non clinicamente rilevanti. Denominazione e identificazione dei farmaci biologici, compresi i farmaci biosimilari Come richiesto dalla normativa europea, tutti i farmaci autorizzati alla commercializzazione devono presentare una denominazione commerciale che può essere rappresentata da un nome di fantasia (brand) oppure dalla denominazione del principio attivo 3 seguita da un marchio o dal 3 La denominazione del principio attivo o INN (International Non Proprietary Name) viene valutata e registrata dall Organizzazione mondiale della sanità (OMS) a seguito della presentazione di una domanda da parte del produttore del medicinale. Rispetto ai farmaci biologici e biotecnologici l OMS adotta un sistema di denominazione formulato da un apposito comitato di esperti dell OMS (INN Expert Group). In particolare, le linee guida OMS lasciano facoltà al produttore di un medicinale biosimilare di proporre un differente INN rispetto al medicinale biologico di riferimento. 7 Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari
8 nome dell'azienda titolare di AIC. Pertanto, ogni farmaco biologico, inclusi i biosimilari, è chiaramente identificabile attraverso una denominazione univoca formalmente approvata dall EMA come parte del processo di autorizzazione. La denominazione commerciale approvata, insieme al numero di lotto, è importante per una chiara identificazione del farmaco ai fini del monitoraggio delle reazioni avverse e per raccogliere le informazioni sul farmaco e quindi garantirne la tracciabilità. Sicurezza dei prodotti biosimilari I medicinali biosimilari devono essere prodotti secondo gli stessi standard qualitativi richiesti per gli altri medicinali (es. prodotti di sintesi chimica e biologici di riferimento). I produttori di farmaci biologici e biosimilari sono tenuti ad istituire, secondo le normative vigenti, un sistema di farmacovigilanza per il monitoraggio della sicurezza del prodotto. Tale sistema di farmacovigilanza è sottoposto a controlli da parte delle Autorità regolatorie che, come per tutti i medicinali, sono tenute a svolgere ispezioni periodiche del prodotto, degli stabilimenti di produzione e del sistema di monitoraggio, sia in fase pre-autorizzativa sia durante la commercializzazione. Ogni azienda è tenuta a presentare, unitamente alla domanda di autorizzazione all'immissione in commercio, un piano di gestione del rischio ( Risk Management Plan ; EU-RMP). L EU-RMP deve esplicitare in dettaglio il sistema di gestione del rischio, descrivendo il profilo di sicurezza del farmaco, tenendo conto anche del profilo di sicurezza noto del corrispondente medicinale di riferimento, e delineare le modalità con cui il produttore continuerà a monitorare la sicurezza e l efficacia del medicinale e le misure che i titolari dell autorizzazione intendono introdurre per prevenire o minimizzare gli eventuali rischi durante l'uso del medicinale, ivi compresa la misurazione dell efficacia nella pratica clinica. L'EU-RMP di un prodotto medicinale biosimilare è un programma specifico di gestione del rischio e deve essere approvato dalle Autorità competenti prima che il farmaco sia commercializzato. Ogni farmaco biosimilare sul mercato ha un EU-RMP in corso, con una sintesi pubblicata nella Relazione di Valutazione Pubblica Europea (EPAR). La nuova normativa in materia di farmacovigilanza (Direttiva 2010/84/EU) prevede che per tutti i prodotti medicinali nel riassunto delle caratteristiche del prodotto e nel foglio illustrativo debba essere incluso un testo standard che incoraggi gli operatori sanitari e i pazienti a segnalare qualsiasi sospetta reazione avversa (Adverse Drug Reaction - ADR) in conformità con i sistemi nazionali di segnalazione spontanea. Per la comunicazione di ADR relativa a tutti i prodotti Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 8
9 biologici, tra i quali anche i biosimilari, la chiara identificazione del farmaco è di particolare importanza. Pertanto, la legislazione europea richiede che in ogni segnalazione di reazioni avverse ad un farmaco biologico, la denominazione del prodotto, così come approvato, e il numero di lotto debbano essere incluse nella relazione di ADR. Inoltre, la menzionata Direttiva 2010/84/EU classifica i medicinali biologici e i biosimilari come priorità per le attività di farmacovigilanza e ne impone l inclusione in una specifica lista di prodotti soggetti a monitoraggi aggiuntivi. In tal senso la presenza, presso l AIFA, della Rete Nazionale di Farmacovigilanza rappresenta uno strumento essenziale di implementazione della recente legislazione europea che consentirà di aumentare la trasparenza, la comunicazione e la fiducia tra le istituzioni e i cittadini per il corretto utilizzo dei farmaci biologici e biosimilari. Infine, sempre la Direttiva 2010/84/EU, dispone che l autorizzazione all'immissione in commercio possa essere subordinata alla condizione di eseguire studi post-autorizzazione di sicurezza (PASS) e/o di efficacia (PAES). Gli studi PASS hanno l obiettivo di identificare, caratterizzare e quantificare un rischio per la sicurezza o confermare il profilo di sicurezza del farmaco, oppure misurare l'efficacia delle misure di gestione dei rischi durante la commercializzazione del farmaco (in questo ambito rientrano, nello specifico, i fenomeni di immunogenicità che rappresentano un problema di sicurezza essenziale di qualsiasi farmaco biologico e che sono obbligatoriamente gestiti nell EU- RMP). Gli studi PAES hanno, invece, l obiettivo di valutare e confermare l efficacia in casi in cui esistano incertezze relative ad alcuni aspetti dell efficacia di un medicinale che possono essere chiarite solo dopo la sua commercializzazione. La Direttiva 2010/84/EU è consultabile sul sito dell EMA al seguente indirizzo link: sting_ jsp&mid=wc0b01ac b3 L'estrapolazione delle indicazioni terapeutiche I farmaci biologici sono spesso autorizzati per più indicazioni terapeutiche. L'estrapolazione delle indicazioni è stata riconosciuta dall EMA, che afferma che: "Nel caso in cui il farmaco originatore sia autorizzato per più di una indicazione, l efficacia e la sicurezza del farmaco biosimilare devono essere confermate o, se necessario, dimostrate separatamente per ogni singola indicazione. In taluni casi può essere possibile estrapolare la somiglianza terapeutica dimostrata in un indicazione ad altre indicazioni autorizzate per il medicinale di riferimento. La giustificazione per Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 9
10 l estrapolazione dovrà tener conto, ad esempio, dell esperienza clinica, dei dati disponibili in letteratura, del meccanismo d azione e dei recettori coinvolti nelle diverse indicazioni. Devono anche essere investigati eventuali problemi di sicurezza in differenti sottopopolazioni. In ogni caso, l azienda produttrice deve giustificare l approccio utilizzato durante lo sviluppo del prodotto consultando l EMA per eventuali chiarimenti di natura scientifica e regolatoria prima dell inizio del programma di sviluppo (EMEA/CHMP/BMWP/42832/2005). La possibilità di estrapolare alcune indicazioni si basa sempre sulla dimostrata comparabilità tra il biosimilare e il prodotto di riferimento rispetto agli standard di qualità, sicurezza ed efficacia nella specifica indicazione raggiunti dal prodotto di riferimento approvato. Solo dopo che siano state dimostrate la comparabilità della qualità e la comparabilità pre-clinica e clinica, è ammissibile che nel dossier di un prodotto biosimilare si faccia riferimento ai dati clinici ottenuti con il prodotto di riferimento e descritti nella letteratura e nei relativi dossier. Va, altresì, sottolineato che il Committee for Medicinal Products for Human Use (CHMP) dell EMA stabilisce caso per caso se le indicazioni multiple possano essere estrapolate sulla base delle evidenze scientifiche derivanti da un esercizio di comparabilità approfondita e in conformità ad opportune giustificazioni scientifiche. Utilizzo di biosimilari nell ambito della Legge n.648/1996 In Italia, la legge n. 648/96 ha previsto, tra l altro, che per il trattamento di una patologia per la quale non sia disponibile una valida alternativa terapeutica possono essere impiegati ed erogati a carico del SSN farmaci autorizzati per altra indicazione terapeutica (utilizzo off-label) per i quali siano disponibili dati di sicurezza ed efficacia raccolti in studi clinici almeno di fase IIprevio parere della Commissione consultiva Tecnico Scientifica (CTS) dell AIFA. I medicinali che acquisiscono parere favorevole dalla CTS vengono inseriti in un apposito elenco, così come stabilito dal Provvedimento del 20 luglio 2000 Istituzione dell'elenco delle specialità medicinali erogabili a totale carico del Servizio Sanitario nazionale ai sensi della legge n.648/96. Il medesimo provvedimento ha indicato come requisiti minimi affinché un farmaco venga inserito in tale elenco e sia dunque a carico del SSN non solo la mancanza di alternative terapeutiche valide, ma anche la disponibilità di dati di sicurezza ed efficacia raccolti in studi clinici almeno di fase II. Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 10
11 Poiché anche i prodotti biologici possono essere utilizzati per l uso off-label, nel caso di un medicinale biosimilare il cui corrispondente medicinale biologico di riferimento sia già stato autorizzato per l utilizzo off-label e sia, quindi, presente nel richiamato elenco l inserimento del biosimilare non è automatico, ma verrà verificato caso per caso dalla CTS, che si riserva la possibilità di effettuare le proprie valutazioni caso per caso sulla base delle evidenze e della letteratura disponibili, dell esperienza clinica e dell eventuale riconducibilità dell azione terapeutica ad un identico meccanismo d azione. Ruolo dei biosimilari nel Sistema sanitario italiano I farmaci biologici rappresentano una risorsa terapeutica essenziale per il trattamento di una varietà di malattie gravi e debilitanti per molte delle quali non erano in passato disponibili opzioni terapeutiche efficaci. Per motivi di sviluppo e produzione del farmaco, questi medicinali sono gravati da costi particolarmente onerosi per il SSN, per il quale la corretta allocazione delle risorse terapeutiche ed economiche rappresenta una sfida costante. In questo scenario i medicinali biosimilari possono svolgere un ruolo nodale offrendo l'opportunità di garantire l'accesso ai farmaci biologici per tutti i pazienti che ne necessitano e contribuendo, nel contempo, alla sostenibilità finanziaria dei sistemi sanitari. La commercializzazione dei farmaci biosimilari può contribuire a migliorare l'accesso ai farmaci in due modi: in primo luogo i biosimilari possono rendere farmaci biologici poco accessibili, perché ad alto costo, più sostenibili e fruibili, innescando meccanismi di competitività dei mercati che determinano riduzione dei prezzi; in secondo luogo, i risparmi generati dall utilizzo dei biosimilari possono contribuire al finanziamento di nuovi farmaci, anche biotecnologici, rendendo sempre più accessibile l'innovazione terapeutica. Il mercato europeo per i biosimilari è rilevante e in espansione, non solo per le aziende produttrici di questi farmaci, ma anche per i sistemi sanitari che sostengono i costi di tali prodotti. Tuttavia è da evidenziare che esistono notevoli differenze tra gli Stati Membri dell UE relativamente sia ai tassi di utilizzo dei biosimilari sia alle politiche di fissazione e regolazione dei prezzi. In Italia le procedure di prezzo e rimborso dei prodotti biologici e dei biosimilari sono le medesime e prevedono una procedura negoziale del prezzo condotta dall AIFA con il produttore (Delibera CIPE 1 febbraio 2001). Nel caso dei biosimilari, per consuetudine e per analogia con i farmaci equivalenti, la negoziazione del prezzo ha come riferimento una riduzione obbligatoria del prezzo, pari ad almeno il 20% rispetto al prezzo del farmaco di riferimento. Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 11
12 I biosimilari come alternativa ai prodotti originatori: la questione della sostituibilità L argomento della sostituibilità, che caratterizza il mondo dei farmaci equivalenti, rappresenta un aspetto importante anche per l affermazione dei medicinali biosimilari. Prima di affrontare in dettaglio l argomento della sostituibilità appare necessario fare chiarezza circa concetti di intercambiabilità e di sostituibilità. Riguardo al concetto di intercambiabilità riferita alla pratica medica si riportano le seguenti definizioni: Secondo la definizione dell'oms è prodotto farmaceutico intercambiabile: un prodotto che si prevede abbia lo stesso effetto clinico di un prodotto comparatore e possa essere sostituito ad esso nella pratica clinica (Ref: WHO Technical Report Series, No. 937, 2006). L intercambiabilità si riferisce alla pratica medica di sostituire un farmaco con un altro equivalente in un determinato contesto clinico su iniziativa o con l'accordo del medico prescrittore (definizione European Generic medicines Association, EGA). L intercambiabilità si riferisce alla pratica medica di sostituire un farmaco con un altro che ha lo stesso profilo beneficio-rischio e che si prevede abbia lo stesso effetto clinico in un determinato contesto clinico su iniziativa o con l'accordo del medico prescrittore. (definizione EFPIA-EBE/EuropaBIO). Al contrario, in ambito esclusivamente statunitense, i termini intercambiabile o intercambiabilità, in riferimento ad un prodotto biologico, indicano che il prodotto biosimilare può essere sostituito al prodotto di riferimento senza l'intervento del medico che ha prescritto quest ultimo." (Biologics Price Competition and Innovation Act, BPCI Act SEC b). In tale contesto, la definizione di intercambiabilità di un biosimilare rispetto al prodotto di riferimento viene, infatti, stabilita, da parte di una commissione della FDA sulla base della documentazione che deve essere presentata rispondendo a specifici criteri definiti a priori. Quindi, una volta definito intercambiabile il prodotto biosimilare, non è necessaria per la sua sostituzione la decisione del clinico sul singolo caso. La sostituibilità fa, invece, riferimento alla pratica di sostituire un farmaco con un altro farmaco, spesso più economico per il Servizio Sanitario o per il paziente che abbia la stessa composizione qualitativa e quantitativa di sostanze attive, la stessa forma farmaceutica e via di somministrazione e sia bioequivalente con il medicinale di riferimento sulla base di appropriati studi di Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 12
13 biodisponibilità. La sostituibilità automatica (degli equivalenti) da parte dei farmacisti si riferisce alla pratica per cui il farmacista ha la facoltà, oppure è tenuto, conformemente a norme nazionali o locali, a dispensare, al posto del medicinale prescritto, un farmaco equivalente e intercambiabile, senza consultare il medico prescrittore. Infine, per quanto riguarda la sostituibilità si possono distinguere,: la sostituibilità primaria, che si riferisce alla pratica medica di iniziare un nuovo trattamento con un prodotto biosimilare (o con un equivalente) piuttosto che con il prodotto originatore di riferimento; la sostituibilità secondaria che si riferisce, invece, alla pratica medica e/o del farmacista di modificare la terapia di un paziente già in trattamento con un farmaco biologico con il suo biosimilare. In merito alla sostituibilità automatica dei biosimilari, la legislazione europea ha affidato alle Autorità nazionali competenti dei differenti stati membri autonomia decisionale e legislativa in materia. Tuttavia, l EMA ha precisato che le raccomandazioni emanate dall EMA sull immissione in commercio del medicinali non comprendono l opportunità o meno di utilizzare un medicinale biosimilare in maniera intercambiabile e che la decisione circa la scelta prescrittiva del medicinale specifico da impiegare, di riferimento piuttosto che biosimilare, debba essere affidata a personale sanitario qualificato (Ref. EMEA/74562/2006 Rev. 1; EMA/837805/2011). In Italia la posizione dell AIFA, chiarisce che i medicinali biologici e biosimilari non possono essere considerati sic et simpliciter alla stregua dei prodotti equivalenti, escludendone quindi la vicendevole sostituibilità terapeutica automatica. Proprio perché i medicinali biologici di riferimento ed i biosimilari sono medicinali simili, ma non identici, l AIFA ha deciso di non includere i medicinali biosimilari nelle liste di trasparenza che consentono la sostituibilità automatica tra prodotti equivalenti. Di conseguenza, la scelta di trattamento con un farmaco biologico di riferimento o con un biosimilare rimane una decisione clinica affidata al medico specialista prescrittore. L AIFA considera, tuttavia, che i biosimilari non solo costituiscono un opzione terapeutica a disposizione dei curanti, ma sono da preferire, qualora costituiscano un vantaggio economico, in particolare per il trattamento dei soggetti naive (che non abbiano avuto precedenti esposizioni terapeutiche o per i quali le precedenti esposizioni in base al giudizio del clinico siano sufficientemente distanti Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 13
14 nel tempo). Inoltre, in considerazione del fatto che il processo di valutazione per la designazione della biosimilarità è condotta dall EMA al massimo livello di conoscenze scientifiche e sulla base di tutte le evidenze disponibili, non sono necessarie ulteriori valutazioni comparative effettuate a livello regionale o locale. Si rappresenta infine che l AIFA si riserva, comunque, di valutare caso per caso l applicabilità dei principi generali enunciati in questo Position Paper, nonché quella di modificare le proprie posizioni sui singoli prodotti e/o sulle singole categorie terapeutiche in relazione al tempo di commercializzazione dei prodotti interessati, alle evidenze scientifiche acquisite, al numero di pazienti trattati nella pratica clinica, agli PSUR presentati all EMA, agli studi PAES e PASS, e alle informazioni estrapolabili da eventuali registri. Conclusioni Lo sviluppo e l utilizzo dei farmaci biosimilari rappresentano un opportunità essenziale per l ottimizzazione dell efficienza dei sistemi sanitari e assistenziali, avendo la potenzialità di soddisfare una crescente domanda di salute, in termini sia di efficacia e di personalizzazione delle terapie sia di sicurezza d impiego. I medicinali biosimilari rappresentano, dunque, uno strumento irrinunciabile per lo sviluppo di un mercato dei biologici competitivo e concorrenziale, necessario alla sostenibilità del sistema sanitario e delle terapie innovative, mantenendo garanzie di sicurezza e qualità per i pazienti e garantendo loro un accesso omogeneo e tempestivo ai farmaci innovativi, pur in un contesto di razionalizzazione della spesa pubblica. Si segnala, infine, che subito prima della pubblicazione di questo Position Paper è stata pubblicata sul sito web dell EMA una revisione della linea guida Draft guideline on similar biological medicinal products (CHMP/437/04 Rev.1) per pubblica consultazione e raccolta di commenti ( Comment invited on draft revised guideline on biosimilars ) di cui si consiglia la lettura al fine di integrare quanto esposto nel presente Position Paper e come aggiornamento della Bibliografia. Bibliografia EMA Procedural advice for users of the Centralised Procedure for Similar Biological Medicinal Products applications -Document September 2012 (EMA/940451/2011). Domande e risposte sui medicinali biosimilari - Q&A 27 September 2012 (EMA/837805/2011) Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 14
15 EMA Concept Paper Revision of the guideline on similar biological medicinal product (CHMP/BMWP/572643/2011) EMA Guideline Similar biological medicinal products (CHMP/437/04) EMA Guideline Comparability of medicinal products containing biotechnology-derived proteins as active substance - Quality issues (CPMP/ICH/5721/03) ICH topic S6 Note for guidance on preclinical safety evaluation of biotechnology-derived pharmaceuticals (CPMP/ICH/302/95) EMA Guideline Similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (EMEA/CHMP/BMWP/42832/2005) EMA Guideline Similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (EMEA/CHMP/BWP/49348/2005) Draft guideline on similar biological medicinal products containing monoclonal antibodies (EMA/CHMP/BMWP/403543/2010) EMA Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo use (EMA/CHMP/BMWP/86289/2010 Rev.2) EMA Guideline Similar biological medicinal products containing recombinant erythropoietins (EMEA/CHMP/BMWP/301636/08) EMA Guideline Non-clinical and clinical development of similar medicinal products containing recombinant interferon alpha (EMEA/CHMP/BMWP/102046/2006) Guideline on evaluation of similar biotherapeutic products (SBPs), WHO 2009 Multisource (Generic) Pharmaceutical Products: Guidelines On Registration Requirements To Establish Interchangeability World Health Organization WHO Technical Report Series, No. 937, 2006 Biologics Price Competition and Innovation Act, BPCI Act SEC b Carson KL. Flexibility The guiding principle for antibody manufacturing. Nat Biotech 2005; 23: Agenzia Italiana del Farmaco Position Paper sui Farmaci Biosimilari 15
16 Commissione Interdisciplinare Biosimilari Task Force Operativa
17 Dal sondaggio AIOM del maggio 2008 II 17% dei rispondenti si dichiara familiare col significato del termine biosimilare il 43% non ne ha mai sentito parlare; Il 70% ritiene che per ogni farmaco ''copia" del biotecnologico originator si debbano effettuare studi di confronto con la molecola originale prima di ricevere l'autorizzazione all'immissione in commercio. Il 50% ritiene che la numerosità della casistica debba essere identica a quella dei farmaci originali. Il 20% è contrario a una decisione che consenta l'impiego del farmaco biosimilare prima dell erogazione di adeguata legislazione in merito (il 20% è favorevole, il 46% "non sa"). CIPOMO Commissione Interdisciplinare Biosimilari
18 Scenario attuale I brevetti di diversi biotecnologici sono scaduti o stanno per scadere creando una imminente disponibilità, anche in Italia, dei biosimilari, che sono farmaci che, come le versioni generiche dei farmaci di sintesi tradiizonali, nascono con il preciso obiettivo di ampliare l offerta e di realizzare risparmi di spesa Le Società e le Associazioni Scientifiche vogliono portare il proprio contributo per approfondire la conoscenza dei biosimilari (potenzialità, profilo di sicurezza), che sono farmaci destinati nel tempo ad assumere un importanza sempre maggiore con previsione al 2020 di circa il 50% di farmaci di natura biotecnologica in commercio (dati Intecontinental Marketing Services). EMEA demanda i singoli Stati Membri della Comunità Europea a esprimersi attraverso provvedimenti normativi specifici. CIPOMO costituisce la Commissione Interdisciplinare Biosimilari, il 12 maggio 2008, in occasione della tavola rotonda di Forlì, per approfondire le problematiche legate alla commercializzazione dei primi biosimilari. CIPOMO Commissione Interdisciplinare Biosimilari
19 Composizione Task Force Operativa Maria Pia Abbracchio Dipartimento di Scienze Farmacologiche, Università degli Studi di Milano Mario Airoldi Struttura Complessa di Oncologia Medica 2, Azienda Ospedaliera Universitaria San Giovanni Battista, Torino Francesco Di Costanzo (Coordinatore) Struttura Complessa di Oncologia Medica, Azienda Ospedaliera Universitaria Careggi, Firenze Salvatore Palazzo Struttura Complessa di Oncologia medica, Azienda Ospedaliera, Cosenza CIPOMO Commissione Interdisciplinare Biosimilari
20 In press CIPOMO Commissione Interdisciplinare Biosimilari
21 Scopi del Documento di Indirizzo Contribuire all inquadramento delle problematiche e delle opportunità connesse all uso di biosimilari, come stimolo alle Autorità regolatorie, ai decisori di spesa e agli stakeholders. Salvaguardare il paziente oncologico, ponendo le basi per la valutazione dell accesso alle migliori opportunità terapeutiche individuali, al minor costo di rischio terapeutico e consentendogli di partecipare in modo consapevole alle scelte terapeutiche. Fornire un contributo all analisi farmaco-economica Costruire un apporto valutativo anche per indicazioni diverse da quelle oncologiche. CIPOMO Commissione Interdisciplinare Biosimilari
22 CIPOMO - Commissione Interdisciplinare Biosimilari Task Force Operativa: metodologia di lavoro Analisi delle evidenze pubblicate e delle legislazioni di altri Stati Membri della CE e internazionali. Evidenziazione delle principali problematiche emerse nell uso clinico dei biosimilari. Condivisione e stesura del documento di indirizzo e presentazione al CIPOMO. CIPOMO Commissione Interdisciplinare Biosimilari
23 CIPOMO - Commissione Interdisciplinare Biosimilari Step Operativi Condivisione ed approvazione del documento presentato dalla task force operativa Messa on-line del documento sul sito CIPOMO, per la raccolta di commenti ed osservazioni da parte dei Soci Implementazione del documento Finalizzazione del documento e divulgazione. CIPOMO Commissione Interdisciplinare Biosimilari
24 FARMACI CHIMICI TRADIZIONALI e FARMACO GENERICO I Farmaci chimici tradizionali sono composti: a basso peso molecolare, a sintesi relativamente semplice e con riproducibilità dei procedimenti produttivi e di analisi a struttura chimica ben definita e facilmente controllabili mediante metodi di analisi standard; con purificazione dai sottoprodotti di reazione basata su procedure standardizzate, costituite da pochi passaggi; con processo produttivo ben definito e facilmente riproducibile. Tutto ciò consente quindi, allo scadere delle coperture brevettuali, di ottenere copie identiche: i "generici, che, oltre alla equivalenza fisicochimica (molecola, formulazione, via di somministrazione, posologia), devono dimostrare una corrispondenza clinica, attraverso studi di farmacocinetica e farmacodinamica - condotti su una casistica clinica limitata - che testimoniano la sovrapponibilità rispetto al farmaco originale ("similarità essenziale"); questa dimostrazione viene assunta a garanzia dell'attività clinica. CIPOMO Commissione Interdisciplinare Biosimilari






25 FARMACO BIOTECNOLOGICO 1- contiene uno o più principi attivi prodotti o derivati a partire da un organismo vivente; 2- si caratterizza per complessa struttura proteica tridimensionale, per elevato peso molecolare - con pesi fino a volte superiori rispetto ai farmaci tradizionali ; 3- prodotto con procedimenti complessi da linee cellulari modificate geneticamente, spesso eterogenei;
 non possono essere \"assimilati\" ai farmaci generici.")
26 A causa della loro complessità strutturale e delle caratteristiche peculiari del processo produttivo (vedi anche oltre), i farmaci biotecnologici non possono essere "assimilati" ai farmaci chimici tradizionali e, di conseguenza, le loro copie (i biosimilari) non possono essere "assimilati" ai farmaci generici. CIPOMO Commissione Interdisciplinare Biosimilari
27 Complessità della produzione di un farmaco biotecnologico Sviluppo della cellula ospite vivente (ceppo produttivo, procarioti, E. coli; o eucarioti, lieviti), non disponibile in commercio, attraverso l'inserimento della sequenza umana di DNA codificante per la proteina desiderata all'interno del genoma di una cellula ospite batterica o eucariote, utilizzando un opportuno vettore; Selezione e costruzione di una banca di cellule costituita da cellule identiche in grado di produrre la stessa proteina umana ricombinante. Ogni banca di cellule è unica, in quanto in ogni laboratorio le linee cellulari vengono donate e crescono generando ceppi diversi. Ogni ceppo produce proteine biotecnologiche simili ma non identiche. Produzione della proteina, attraverso la coltivazione delle cellule su larga scala (l'unicità della banca di cellule si trasferisce alla fase di produzione). Purificazione: è una fase costosa, richiede notevole expertise e costituisce l'elemento di massima criticità per l'attività clinica. Proteine naturali, zuccheri, acidi grassi, elettroliti ed elementi più dannosi quali virus, endotossine, prioni sono fra i prodotti cellulari indesiderati e i contaminanti da eliminare attraverso la purificazione. Una delle maggiori difficoltà è rappresentata dal fatto che, diversamente dai farmaci tradizionali, non sempre è possibile prevedere la natura dei contaminanti, i quali spesso costituiscono la quota percentualmente più elevata dei prodotti di reazione di tipo allergico. Analisi: si fondano su saggi scarsamente sensibili (rispetto a quelli applicati a un farmaco tradizionale), quali l'elettroforesi e le analisi biologiche; definiscono l'uniformità come struttura e potenza, ma discriminano scarsamente in termini di efficacia e sicurezza. Formulazione: la fase di trasformazione del principio attivo in forma farmaceutica somministrabile. La semplice sostituzione di un ingrediente della formulazione con un nuovo ingrediente in grado di interferire con la struttura e/o la biodisponibilità della proteina ricombinante può comportare gravi reazioni indesiderate. Conservazione e manipolazione: condizioni adeguate di conservazione e di manipolazione sono essenziali per mantenere le proprietà biologiche e terapeutiche. I farmaci biotecnologici sono più sensibili dei farmaci tradizionali; l'agitazione del prodotto, l'interruzione della catena del freddo sono solo esempi di CIPOMOdella Commissione situazioni che possono portare alla degradazione proteina con perdita di attività biologica desiderata, o Biosimilari all'aggregazione di particelle proteiche,interdisciplinare con gravi rischi di immunogenicità.
28 Riproducibilità della produzione del farmaco biotecnologico Una banca cellulare non può essere identica a un'altra. Le modalità e le condizioni di produzione, di purificazione, di conservazione e di manipolazione possono modificare le proprietà biologiche e l'attività clinica; la produzione ottenuta a partire da organismi viventi crea prodotti finali diversi rispetto alla loro struttura tridimensionale, al grado di glicosilazione e alle percentuali delle differenti isoforme: qualsiasi variazione può avere un profondo impatto sull'attività biologica del prodotto finale e influenzare efficacia e sicurezza; le possibili differenze di attività biologica potrebbero non essere evidenziate in modo completo fino a quando non sia stata stabilita un'effettiva esperienza d'uso. Pertanto i biosimilari, non possono essere considerati copie conformi dell originator come avviene per le piccole molecole attraverso i generici. CIPOMO Commissione Interdisciplinare Biosimilari
 simile significa infatti analogo o affine nell aspetto o nei caratteri : la sua adozione pertanto avrebbe potuto creare equivoci, rispetto all evidenza che si")
29 SEMANTICA In linea con la denominazione fornita da EMEA (biosimilar), la scelta di impiegare il termine composto bio-similare e non l aggettivo simile non è casuale. Sec. il Dizionario Italiano Devoto-Oli (2007) simile significa infatti analogo o affine nell aspetto o nei caratteri : la sua adozione pertanto avrebbe potuto creare equivoci, rispetto all evidenza che si tratta di un prodotto ben diverso dal biotecnologico originario. La scelta e l adozione dell aggettivo similare - che significa appartenente alla stessa specie (ibidem) - sta a indicare invece in modo incontrovertibile e inequivocabile che si tratta di un prodotto con caratteristiche ben diverse dal biotecnologico originario, al quale è legato dall appartenenza alla stessa categoria farmacologica. CIPOMO Commissione Interdisciplinare Biosimilari
30 Sintesi e definizione di biosimilare: Medicinale biotecnologico che fa riferimento a un farmaco già esistente e che viene sottoposto alle Autorità regolatorie per l'autorizzazione alla commercializzazione dopo la scadenza del brevetto del farmaco biotecnologico originatore. Si tratta di un farmaco similare, cioè non identico al farmaco originale, in quanto le caratteristiche del farmaco biotecnologico e della sua produzione non consentono l'ottenimento di un prodotto "copia" esattamente identico. Ogni farmaco biosimilare è un farmaco nuovo, ma non necessariamente innovativo, diverso sia dal farmaco originale che dagli altri farmaci biosimilari. Trattandosi di un farmaco nuovo, deve essere sottoposto a un'attenta farmacovigilanza e deve essere pienamente caratterizzato in clinica. the process is the product CIPOMO Commissione Interdisciplinare Biosimilari


31 CIPOMO Commissione Interdisciplinare Biosimilari

32 CIPOMO Commissione Interdisciplinare Biosimilari
33 GLI ASPETTI REGOLATORI EUROPEI E ITALIANI L'Unione Europea ha già approvato cinque biosimilari (di cui due soli già in distribuzione in alcuni Paesi). I primi due biosimilari sono stati approvati dall'emea in aprile e maggio 2006 e "lanciati" in Germania e Austria: sono prodotti biosimilari della somatropina per disturbi della crescita in pediatria e come terapia di sostituzione ormonale per gli adulti con marcato deficit dell'ormone della crescita. Nel giugno 2007 il CHMP (Comitato EMEA) ha dato parere positivo a tre prodotti biosimilari deteritropoietina alfa per il trattamento dell'anemia. Eritropoietine biosimilari sono già disponibili in Croazia, Romania e altri Paesi UE, ma senza che vi sia una regolamentazione unica di tali prodotti. In Italia, con determinazione dell'agenzia Italiana del Farmaco, lo scorso febbraio (Gazzetta Ufficiale n. 79 del , Supplemento Ordinario n. 78, pag. 72) sono stati assegnati regime di rimborsabilità e prezzo di vendita a una delle tre epoietine biosimilari sopra considerate, e cioè la specialità medicinale BINOCRIT di Sandoz. Non risultano provvedimenti analoghi per le epoietine Hexal e Abseamed. L'AUTORIZZAZIONE ALL'IMMISSIONE IN COMMERCIO DEI BIOSIMILARI IN EUROPA Come per ogni farmaco, anche per i biosimilari la commercializzazione deve essere autorizzata da un ente regolatorio. In Europa è I'EMEA che effettua una valutazione scientifica di efficacia, sicurezza e qualità del prodotto, con l'obiettivo di rendere disponibile una nuova opzione terapeutica. In anticipo sui tempi rispetto alle Autorità statunitensi - dove ancora non sono scaduti brevetti - I'EMEA ha realizzato Linee Guida che consentono di valutare e di autorizzare l'immissione in commercio di farmaci biosimili (Tab. 1). Questa legislazione prevede un processo di approvazione differente da quello dei farmaci generici e in parte sarà differenziato sulla base di ogni biosimile considerato. Nello sviluppo di una Linea Guida, gli aspetti terminologici sono di estrema importanza: la discussione si è centrata su comparabilità e similarità: per comparabilità si intende la costanza delle caratteristiche di due lotti prodotti dalla stessa azienda; per similarità si intende la "rassomiglianza" di un biosimilare al farmaco originale di riferimento (2).
 ma è demandata a ogni Stato membro.")
34 LA POSIZIONE DELL'EMEA La definizione delle regole amministrative locali e delle misure educazionali per una gestione appropriata non rientra infatti tra i compiti delle Autorità regolatone centrali (EMEA) ma è demandata a ogni Stato membro. Dalle Linee Guida promulgate emerge la posizione dell'emea: 1- i biosimilari non sono uguali ai farmaci biotecnologici originatori; 2- vengono tuttavia approvati per l'immissione in commercio sulla base di uno studio di non inferiorità sulla prima delle indicazioni approvate; 3- il programma clinico registrativo imitato deve essere integrato con l'impegno del produttore a condurre una farmacovigilanza post-marketing efficace. E su questa base la Francia per prima, seguita da altri Stati Membri Europei - Germania, Slovenia, Spagna e Svezia - hanno già normato la materia recependo le Linee Guida europee. In particolare Francia e Spagna vietano l'inserimento dei biosimilari nella lista dei farmaci sostituibili, senza CIPOMO Commissione una richiesta esplicita del Interdisciplinare Biosimilari prescrittore.
35 In Italia, le Autorità non hanno ancora emesso disposizioni al riguardo, anche se è stato presentato al Senato nel dicembre 2007 un progetto su interventi urgenti in materia di spesa sanitaria e nuove disposizioni in materia di farmaci biosimilari. A questo riguardo, va ricordato che già nel 2006, la XII Commissione permanente Affari sociali aveva avuto modo di esprimersi affermando: "con riferimento, infine, ai medicinali prodotti da biotecnologie, appare opportuno sostituire il termine "biogenerici", ovunque ricorra, con il termine "biosimilari", in modo da uniformare le espressioni comprese nello schema di decreto legislativo con quelle già utili77ate nel diritto comunitario". Un parere è quello espresso nel febbraio 2008 dal Consiglio di Stato alla Regione Molise, che aveva chiesto chiarimenti su come comportarsi in caso di acquisto dei medicinali ottenuti da proteine o anticorpi monoclonali: l'equivalenza terapeutica non vale per i farmaci biotecnologici (una posizione in linea con quella dell'emea). Per questo parere, il Consiglio di Stato è ricorso al Ministero della Salute, che a sua volta ha interpellato l'istituto Superiore della Sanità e l'agenzia Italiana del Farmaco. Molte regioni hanno risparmiato decine di milioni di euro dando per acquisito il principio di equivalenza per queste cure. Per l'aifa però i "processi produttivi alla base delle cure biotecnologiche rendono molto complessa la messa a punto di farmaci biosimilari". In modo ufficioso, ma autorevole, in occasione di un convegno AIOM tenuto a Roma nel maggio 2008 su "Farmaci biotecnologici e similari", il Direttore dell'aifa ha ribadito il principio della non sostituibilità per alcune categorie di farmaci - compresi i biotecnologici - principio che ribadisce il ruolo chiave del prescrittore. CIPOMO Commissione Interdisciplinare Biosimilari
36 CONCLUSIONI L'imminente disponibilità dei biosimilari - farmaci nuovi, diversi dai biotecnologici di riferimento - vede una forte attenzione delle Società e delle Associazioni Scientifiche, tese a contribuire alla conoscenza dei biosimilari, delle loro potenzialità e del loro profilo di sicurezza, con l'intento di definire una loro appropriata collocazione nell'armamentario terapeutico e di evitare qualsiasi allarmismo e l'insorgenza di emergenze sanitarie. In quest'ottica, la Task Force Operativa, della Commissione Interdisciplinare Biosimilari, CIPOMO, auspica: 4.La definizione di un tavolo di confronto con le istituzioni centrali e regionali, al quale le Associazioni professionali e le Società Scientifiche (CIPOMO, AIOM, SIF, SIFO) possano contribuire in qualità di interlocutori scientifici in modo da partecipare alla definizione della governance clinica. 5.Una decisione centrale da parte dell'aifa, al fine di evitare situazioni diversificate a livello regionale. 6.Un allineamento nell'iter legislativo di approvazione all'immissione in commercio e nelle modalità prescrittive alle scelte già operate da altri Paesi europei (Francia, Germania) che già vietano l'inserimento dei biosimilari nella lista dei farmaci sostituibili senza una richiesta esplicita del prescrittore. 7.Una corretta ed efficace comunicazione istituzionale a tutti gli attori del sistema, predisposta con le Associazioni dei pazienti e le Associazioni del Consumatori. CIPOMO Commissione Interdisciplinare Biosimilari

37 CIPOMO Commissione Interdisciplinare Biosimilari

38 CIPOMO Commissione Interdisciplinare Biosimilari
39 La lettera a) dell art. 1 introduce l esclusione dell equivalenza terapeutica per i farmaci biosimilari e originali come anche quella tra biosimilari diversi in una stessa classe di appartenenza. La lettera b) dell art. 1 specifica che e` il medico che deve avere la piena responsabilità sulla scelta tra farmaco biosimilare o biologico e biotecnologico (4bis) e introduce la garanzia che nei processi d acquisto di farmaci nelle strutture sanitarie venga rispettato il principio di non equivalenza terapeutica tra farmaci biosimilari e originali come anche tra quelli biologici e biotecnologici appartenenti alla stessa classe (4-ter). CIPOMO Commissione Interdisciplinare Biosimilari

40 16/03/2010 CIPOMO Commissione Interdisciplinare Biosimilari




41 Linee guida AIFA e SIF per l uso dei Biosimilari Prof. Giovambattista De Sarro Dipartimento di Scienze della Salute Università degli Studi Magna Graecia di Catanzaro
42 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi nell industria farmaceutica NO Attualmente Precedenti 2 anni Da oltre 2 a 5 anni precedenti Oltre 5 anni precedenti (facoltativo) Interessi diretti: Impiego in una società Consulenza per una società Consulente strategico per una società Interessi finanziari Titolarità di un brevetto x x x x x Interessi indiretti: Sperimentatore principale x Sperimentatore x X Sovvenzioni o altri fondi finanziari x X * Giovambattista De Sarro, secondo il regolamento sul Conflitto di Interessi approvato dal CdA AIFA in data e pubblicato sulla Gazzetta Ufficiale del in accordo con la policy 0044 EMA/513078/2010 sulla gestione del conflitto di interessi dei membri dei Comitati Scientifici e degli esperti. N.B. Per questo intervento non ricevo alcun compenso
43 Lo sviluppo e l utilizzo dei farmaci biosimilari rappresentano un opportunità essenziale per l ottimizzazione della efficienza dei sistemi sanitari e assistenziali, in termini di efficacia, personalizzazione delle terapie e sicurezza d impiego




44 Get back to lab! Translational Medicine ( from bench to bedside, and back!)

45 RICERCA TRASLAZIONALE Studi scientifici di base che definiscano gli effetti biologici dei trattamenti nell uomo; indagini nell uomo che delineino la biologia del disturbo e forniscano il fondamento scientifico per lo sviluppo o il miglioramento di nuove terapie; studi non clinici o animali condotti con lo scopo di migliorare le terapie nella clinica; adeguato sviluppo di un prodotto ad uso clinico nelle varie fasi del processo di studio

46 I farmaci Biosimilari sono medicinali biologici autorizzati dall Agenzia Europea dei Medicinali simili per qualità, efficacia e sicurezza al prodotto biologico di riferimento. La disponibilità dei prodotti Biosimilari rappresenta un fattore importante per il mantenimento della sostenibilità economica dei servizi sanitari nel prossimo futuro.

47 FARMACI BIOTECNOLOGICI: DEFINIZIONE Farmaci progettati mediante la tecnologia del DNA ricombinante.


 l insulina si purificava dal pancreas")
48 Farmaci Biotecnologici Nel 1982 venne approvato il primo prodotto biotecnologico ottenuto ingegnerizzando un sistema vivente (batterico) : insulina umana Anche l ormone umano della crescita è stato rapidamente ingegnerizzato. Prima ( ) l insulina si purificava dal pancreas di suini e bovini. Precedentemente estratto dai cadaveri.

49 Dimensions



50 Farmaci Biotecnologici La maggior parte dei farmaci biotech sono proteine altamente complesse, macromolecole che crescono in sistemi viventi, alcune delle quali costituite da numerosi componenti. Aspirina: Peso molecolare 180 Interferone beta: peso molecolare
51 Farmaci Biotecnologici

52 Farmaci Biosimilari: definizione Farmaci simili a medicinali biotecnologici già in commercio il cui brevetto è scaduto



53 Ad oggi, a livello mondiale, milioni di pazienti hanno già beneficiato dei medicinali biologici approvati per la cura o la prevenzione di molte malattie quali tumori, malattie infiammatorie, autoimmuni, neurologiche e degenerative
54 AIFA POSITION L AIFA ha ritenuto indispensabile fornire un documento il cui obiettivo è quello di promuovere la conoscenza e l utilizzo dei biosimilari relativamente ai seguenti aspetti: Definizione e principali criteri di caratterizzazione dei medicinali biologici e biosimilari; Inquadramento delle normative regolatorie vigenti in EU in merito ai medicinali biosimilari; Ruolo dei biosimilari nella sostenibilità economica del servizio sanitario nazionale (SSN).
 simili per qualità, efficacia e sicurezza al prodotto biologico di")
55 Il concetto di medicinale biologico simile è stato introdotto nella legislazione dell'unione Europea dalla direttiva 2001/83/CE e successive modificazioni I farmaci biosimilari sono, quindi, medicinali biologici autorizzati dall Agenzia Europea dei Medicinali (European Medicines Agency EMA) simili per qualità, efficacia e sicurezza al prodotto biologico di riferimento.
56 Nel settembre 2012 l EMA ha diffuso un documento in cui : Per medicinale biosimilare si intende un medicinale sviluppato in modo da risultare simile a un medicinale biologico che è già stato autorizzato (il così detto medicinale di riferimento ). Come il medicinale di riferimento, il biosimilare presenta un certo grado di variabilità naturale. Viene approvato quando è stato dimostrato che tale variabilità naturale ed eventuali differenze rispetto al medicinale di riferimento non influiscono sulla sicurezza o sull efficacia. (Questions and Answers on biosimilar medicines EMA/837805/2011 del 27 settembre 2012)
.")
57 Farmaci Biosimilari L iter registrativo di un prodotto biosimilare è molto differente da quello dei farmaci equivalenti di medicinali non biologici, per i quali è generalmente sufficiente presentare i risultati degli studi di bioequivalenza (Dir. 2003/63/EC, Ann I, P.II). La dimostrazione di bioequivalenza non basta per qualificare un biosimilare sono necessari studi di comparabilità (comparability) sia per il principio attivo che per il medicinale finito fra l originatore e il biosimilare. Chi produce il biosimilare non ha la disponibilità né dei dati dell originatore, né della materia prima. Può analizzare solo il prodotto finito, e su questa base deve costruire la sua comparability.
58 La normativa richiede che il programma di ricerca e sviluppo sia volto a dimostrare la biosimilarità intesa come la comparabilità tra un biosimilare ed il suo prodotto di riferimento, attraverso l esercizio di comparabilità OVVERO l insieme di procedure di confronto graduale (stepwise) che inizia con studi di comparabilità fisico-chimiche e biologiche, e prosegue con la valutazione della comparabilità di studi non clinici comparativi e clinici comparativi per la valutazione dell efficacia e della sicurezza, includendo lo studio dell immunogenicità. L obiettivo primario dell esercizio di comparabilità è la dimostrazione della similarità ( similarity throughout )

59 Substitution
60 Riguardo al concetto di intercambiabilità riferita alla pratica medica si riportano le seguenti definizioni: Secondo l'oms è prodotto farmaceutico intercambiabile: un prodotto che si prevede abbia lo stesso effetto clinico di un prodotto comparatore e possa essere sostituito ad esso nella pratica clinica (Ref: WHO Technical Report Series, No. 937, 2006). L intercambiabilità si riferisce alla pratica medica di sostituire un farmaco con un altro equivalente in un determinato contesto clinico su iniziativa o con l'accordo del medico prescrittore (definizione European Generic medicines Association, EGA). L intercambiabilità si riferisce alla pratica medica di sostituire un farmaco con un altro che ha lo stesso profilo beneficio-rischio e che si prevede abbia lo stesso effetto clinico in un determinato contesto clinico su iniziativa o con l'accordo del medico prescrittore. (definizione EFPIA-EBE/EuropaBIO).
.")
61 I produttori di farmaci biologici e biosimilari sono tenuti ad istituire, secondo le normative vigenti, un sistema di farmacovigilanza per il monitoraggio della sicurezza del prodotto. Ogni azienda è tenuta a presentare, unitamente alla domanda di autorizzazione all'immissione in commercio, un piano di gestione del rischio ( Risk Management Plan ; EU-RMP). Ogni farmaco biosimilare sul mercato ha un EU-RMP in corso, con una sintesi pubblicata nella Relazione di Valutazione Pubblica Europea (EPAR)
62 La legislazione europea richiede che in ogni segnalazione di reazioni avverse ad un farmaco biologico, la denominazione del prodotto, così come approvato, e il numero di lotto debbano essere incluse nella relazione di ADR Inoltre, la menzionata Direttiva 2010/84/EU classifica i medicinali biologici e i biosimilari come priorità per le attività di farmacovigilanza e ne impone l inclusione in una specifica lista di prodotti soggetti a monitoraggi aggiuntivi. la Direttiva 2010/84/EU, dispone che l autorizzazione all'immissione in commercio possa essere subordinata alla condizione di eseguire studi post-autorizzazione di sicurezza (PASS) e/o di efficacia (PAES).
63 Utilizzo di biosimilari nell ambito della Legge n.648/1996 Per il trattamento di una patologia per la quale non sia disponibile una valida alternativa terapeutica possono essere impiegati ed erogati a carico del SSN farmaci autorizzati per altra indicazione terapeutica (utilizzo off-label) per i quali siano disponibili dati di sicurezza ed efficacia raccolti in studi clinici almeno di fase II previo parere della Commissione consultiva Tecnico Scientifica (CTS) dell AIFA.
64 L AIFA considera che i biosimilari non solo costituiscono un opzione terapeutica a disposizione dei curanti, ma sono da preferire, qualora costituiscano un vantaggio economico, in particolare per il trattamento dei soggetti naive
65 l AIFA, chiarisce che i medicinali biologici e biosimilari non possono essere considerati sic et simpliciter alla stregua dei prodotti equivalenti, escludendone quindi la vicendevole sostituibilità terapeutica automatica. i medicinali biologici di riferimento ed i biosimilari sono medicinali simili, ma non identici, l AIFA ha deciso di non includere i medicinali biosimilari nelle liste di trasparenza che consentono la sostituibilità automatica tra prodotti equivalenti. Di conseguenza, la scelta di trattamento con un farmaco biologico di riferimento o con un biosimilare rimane una decisione clinica affidata al medico specialista prescrittore.
66 L'esercizio di comparabilità è quindi basato su un robusto confronto testa a testa tra il biosimilare e il medicinale di riferimento secondo specifici standard di qualità, sicurezza ed efficacia, avendo definito a priori le differenze ritenute accettabili in quanto non clinicamente rilevanti.
67 Trattamenti a base di infliximab, etanercept e adalimumab sono efficaci nell artrite associata alla psoriasi (Mease et al., 2004; Antoni et al., 2005; Mease et al., 2005). I registri nazionali sugli anti-tnf hanno evidenziato che, nei pazienti con artrite reumatoide, sono efficaci e ben tollerati. Tuttavia per infliximab, adalimumab ed etanercept le rispettive percentuali di risposta terapeutica, remissione della malattia e aderenza al trattamento farmacologico non sono risultate sovrapponibili (Hetland et al., 2010; Greenberg et al.,2012). SIF POSITION PAPER Il 50% circa dei pazienti, che hanno inizialmente risposto al trattamento con un farmaco anti-tnf, interrompono la terapia dopo circa 3-4 anni, a causa di effetti avversi o perdita dell efficacia. (Du Pan et al. 2009; Hetland et al., 2010; Soliman et al., 2011)

68
69
70
71

72

73

74

75 Migliorerà le possibilità prescrittive nel nostro Paese?
Commissione Interdisciplinare Biosimilari Task Force Operativa
 Commissione Task Force Operativa Dal sondaggio AIOM del maggio 2008 II 17% dei rispondenti si dichiara familiare col significato del termine biosimilare il 43% non ne ha mai sentito parlare; Il 70% ritiene
Commissione Task Force Operativa Dal sondaggio AIOM del maggio 2008 II 17% dei rispondenti si dichiara familiare col significato del termine biosimilare il 43% non ne ha mai sentito parlare; Il 70% ritiene
Biosimilare: Normativa di riferimento
 214 A cura di Laura Ricci Area Legislazione SIFO sifo.legislazione@gmail.com Biosimilare: Normativa di riferimento DEFINIZIONE La definizione di farmaco biologico e biosimilare, più volte ripresa e precisata,
214 A cura di Laura Ricci Area Legislazione SIFO sifo.legislazione@gmail.com Biosimilare: Normativa di riferimento DEFINIZIONE La definizione di farmaco biologico e biosimilare, più volte ripresa e precisata,
Linee guida AIFA e SIF per l uso dei Biosimilari
 Linee guida AIFA e SIF per l uso dei Biosimilari Prof. Giovambattista De Sarro Dipartimento di Scienze della Salute Università degli Studi Magna Graecia di Catanzaro Dichiarazione di trasparenza/interessi*
Linee guida AIFA e SIF per l uso dei Biosimilari Prof. Giovambattista De Sarro Dipartimento di Scienze della Salute Università degli Studi Magna Graecia di Catanzaro Dichiarazione di trasparenza/interessi*
Il nuovo Position Paper AIFA sui farmaci biosimilari. Mario
 Il nuovo Position Paper AIFA sui farmaci biosimilari Mario Melazzini m.melazzini@aifa.gov.it @mmelazzini Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
Il nuovo Position Paper AIFA sui farmaci biosimilari Mario Melazzini m.melazzini@aifa.gov.it @mmelazzini Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
Franco Silvestris UNIVERSITÀ DEGLI STUDI DI BARI
 UNIVERSITÀ DEGLI STUDI DI BARI Dipartimento di Scienze Biomediche e Oncologia Umana Sezione di Medicina Interna e Oncologia Clinica U.O.C. Oncologia Medica Universitaria Direttore: Franco Silvestris Franco
UNIVERSITÀ DEGLI STUDI DI BARI Dipartimento di Scienze Biomediche e Oncologia Umana Sezione di Medicina Interna e Oncologia Clinica U.O.C. Oncologia Medica Universitaria Direttore: Franco Silvestris Franco
SECONDO CONCEPT PAPER AIFA SUI FARMACI BIOSIMILARI
 15 giugno 2016 SECONDO CONCEPT PAPER AIFA SUI FARMACI BIOSIMILARI Preparazione del Draft 06.07.2012 Approvazione del Draft e rilascio per la consultazione pubblica 25.07.2012 Inizio della consultazione
15 giugno 2016 SECONDO CONCEPT PAPER AIFA SUI FARMACI BIOSIMILARI Preparazione del Draft 06.07.2012 Approvazione del Draft e rilascio per la consultazione pubblica 25.07.2012 Inizio della consultazione
Biosimilari: Opportunità o Cautela?
 XVII Congresso Nazionale CIPOMO NH Hotel V. Vittorio Veneto,Roma Biosimilari: Opportunità o Cautela? Francesco Di Costanzo SC Oncologia Medica Azienda Ospedaliro Universitaria Careggi Firenze Roma,20-22
XVII Congresso Nazionale CIPOMO NH Hotel V. Vittorio Veneto,Roma Biosimilari: Opportunità o Cautela? Francesco Di Costanzo SC Oncologia Medica Azienda Ospedaliro Universitaria Careggi Firenze Roma,20-22
I BIOSIMILARI TRA PROFILI BREVETTUALI E REGOLATORI 30 OTTOBRE 2015 PARMA MARINA MAURO
 I BIOSIMILARI TRA PROFILI BREVETTUALI E REGOLATORI 30 OTTOBRE 2015 PARMA MARINA MAURO INTRODUZIONE I farmaci I farmaci generici Bioequivalenza I farmaci generici equivalenti I farmaci biologici Biosimilarità
I BIOSIMILARI TRA PROFILI BREVETTUALI E REGOLATORI 30 OTTOBRE 2015 PARMA MARINA MAURO INTRODUZIONE I farmaci I farmaci generici Bioequivalenza I farmaci generici equivalenti I farmaci biologici Biosimilarità
Dr. Andrea M. Machiavelli Direttore U.O.C. Farmacia ASST Cremona
 Dr. Andrea M. Machiavelli Direttore U.O.C. Farmacia ASST Cremona Per le comunicazioni di ADR dei prodotti biologici, inclusi i biosimilari, la chiara identificazione del farmaco è di particolare
Dr. Andrea M. Machiavelli Direttore U.O.C. Farmacia ASST Cremona Per le comunicazioni di ADR dei prodotti biologici, inclusi i biosimilari, la chiara identificazione del farmaco è di particolare
FARMACI BIOSIMILARI IN ONCOLOGIA
 FARMACI BIOSIMILARI IN ONCOLOGIA Opuscolo per cittadini e pazienti Ottobre 2018 Associazione Italiana di Oncologia Medica Associazione Italiana di Oncologia Medica In questo opuscolo troverà informazioni,
FARMACI BIOSIMILARI IN ONCOLOGIA Opuscolo per cittadini e pazienti Ottobre 2018 Associazione Italiana di Oncologia Medica Associazione Italiana di Oncologia Medica In questo opuscolo troverà informazioni,
MEDICINALI BIOEQUIVALENTI E BIOSIMILARI. Dipartimento Farmaceutico Interaziendale Azienda Ospedaliero Universitaria di Ferrara
 MEDICINALI BIOEQUIVALENTI E BIOSIMILARI Dipartimento Farmaceutico Interaziendale Azienda Ospedaliero Universitaria di Ferrara FARMACO GENERICO e EQUIVALENTE Con la Legge 28/12/1995 art. 130 comma 3, il
MEDICINALI BIOEQUIVALENTI E BIOSIMILARI Dipartimento Farmaceutico Interaziendale Azienda Ospedaliero Universitaria di Ferrara FARMACO GENERICO e EQUIVALENTE Con la Legge 28/12/1995 art. 130 comma 3, il
AIFA POSITION PAPER I FARMACI BIOSIMILARI
 AIFA POSITION PAPER I FARMACI BIOSIMILARI Preparazione del Draft 06.07.2012 Presentazione del Draft e rilascio per la consultazione pubblica 25.07.2012 Inizio della consultazione pubblica 1.08. 2012 Fine
AIFA POSITION PAPER I FARMACI BIOSIMILARI Preparazione del Draft 06.07.2012 Presentazione del Draft e rilascio per la consultazione pubblica 25.07.2012 Inizio della consultazione pubblica 1.08. 2012 Fine
DELIBERAZIONE N. 20/5 DEL
 Oggetto: Direttive alle Aziende Sanitarie per l espletamento delle gare per l approvvigionamento di farmaci ed emoderivati relativamente ai farmaci biosimilari. L Assessore dell Igiene e Sanità e dell
Oggetto: Direttive alle Aziende Sanitarie per l espletamento delle gare per l approvvigionamento di farmaci ed emoderivati relativamente ai farmaci biosimilari. L Assessore dell Igiene e Sanità e dell
Farmaci Biosimilari. Un alleanza tra Farmacisti e Reumatologi Per la Sostenibilità del SSR. 13 gennaio 2017 Palazzo Pirelli
 Farmaci Biosimilari Un alleanza tra Farmacisti e Reumatologi Per la Sostenibilità del SSR 13 gennaio 2017 Palazzo Pirelli Cosa Chiedono il SSN e il SSR? Massimo Medaglia Direzione Generale Welfare Regione
Farmaci Biosimilari Un alleanza tra Farmacisti e Reumatologi Per la Sostenibilità del SSR 13 gennaio 2017 Palazzo Pirelli Cosa Chiedono il SSN e il SSR? Massimo Medaglia Direzione Generale Welfare Regione
Secondo Position Paper AIFA sui Farmaci Biosimilari
 Secondo Position Paper AIFA sui Farmaci Biosimilari Indice Premessa 5 Introduzione 6 I medicinali di origine biologica 7 I medicinali biosimilari 9 Normativa vigente in UE in merito ai medicinali biosimilari
Secondo Position Paper AIFA sui Farmaci Biosimilari Indice Premessa 5 Introduzione 6 I medicinali di origine biologica 7 I medicinali biosimilari 9 Normativa vigente in UE in merito ai medicinali biosimilari
Relatore: Dott.ssa Elisabetta Pasi - Servizio Assistenza Territoriale, Direzione Generale Sanità e Politiche Sociali- Regione Emilia-Romagna
 Le decisioni pratiche tra normative, linee guida e raccomandazioni, il position paper di AIFA, l intercambiabilità e la sostituibilità di originator e biosimilari Relatore: Dott.ssa Elisabetta Pasi - Servizio
Le decisioni pratiche tra normative, linee guida e raccomandazioni, il position paper di AIFA, l intercambiabilità e la sostituibilità di originator e biosimilari Relatore: Dott.ssa Elisabetta Pasi - Servizio
Uso dei Biosimilari e sostenibilità: Il Position Paper dell AIFA Simona Montilla
 Uso dei Biosimilari e sostenibilità: Il Position Paper dell AIFA Simona Montilla 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
Uso dei Biosimilari e sostenibilità: Il Position Paper dell AIFA Simona Montilla 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
TITOLO III Immissione in commercio
 TITOLO III Immissione in commercio Art.6 (Estensione ed effetti dell autorizzazione) 1. Nessun medicinale può essere immesso in commercio sul territorio nazionale senza aver ottenuto un'autorizzazione
TITOLO III Immissione in commercio Art.6 (Estensione ed effetti dell autorizzazione) 1. Nessun medicinale può essere immesso in commercio sul territorio nazionale senza aver ottenuto un'autorizzazione
RICERCA E SVILUPPO DI NUOVI FARMACI
 RICERCA E SVILUPPO DI NUOVI FARMACI In passato le scoperte dei farmaci erano occasionali. Si trattava di sostanze di derivazione vegetale o animale STRATEGIE DI RICERCA: Quali sono i fattori che influenzano
RICERCA E SVILUPPO DI NUOVI FARMACI In passato le scoperte dei farmaci erano occasionali. Si trattava di sostanze di derivazione vegetale o animale STRATEGIE DI RICERCA: Quali sono i fattori che influenzano
I biosimilari fra presente e futuro
 ANALISI DELLA PRESCRIZIONE FARMACEUTICA IN ITALIA presentazione del Rapporto Nazionale OsMed 2010 7 luglio 2011 aula Pocchiari I biosimilari fra presente e futuro Anna Maria Marata Lucia Magnano Che cos
ANALISI DELLA PRESCRIZIONE FARMACEUTICA IN ITALIA presentazione del Rapporto Nazionale OsMed 2010 7 luglio 2011 aula Pocchiari I biosimilari fra presente e futuro Anna Maria Marata Lucia Magnano Che cos
STRUTTURA DEL DOSSIER (Allegato 1 del Decreto del Presidente dell'istituto superiore di sanità del 26 aprile 2002)
") STRUTTURA DEL DOSSIER (Allegato 1 del Decreto del Presidente dell'istituto superiore di sanità del 26 aprile 2002) Indirizzo La domanda, così come ogni altra corrispondenza successiva, dovrà essere inviata
STRUTTURA DEL DOSSIER (Allegato 1 del Decreto del Presidente dell'istituto superiore di sanità del 26 aprile 2002) Indirizzo La domanda, così come ogni altra corrispondenza successiva, dovrà essere inviata
A luglio 2012 sono entrate in vigore due nuove disposizioni in tema di sicurezza dei medicinali, dopo più di 10 anni dall ultima normativa europea in
 La normativa europea in materia di farmacovigilanza è stata modificata con l adozione nel 2010, del Regolamento UE 1235/2010, la cui applicazione è operativa dal 2 luglio 2012, e della Direttiva 2010/84/UE,
La normativa europea in materia di farmacovigilanza è stata modificata con l adozione nel 2010, del Regolamento UE 1235/2010, la cui applicazione è operativa dal 2 luglio 2012, e della Direttiva 2010/84/UE,
Lettori: Diffusione: Dir. Resp.: Roberto Napoletano. 06-APR-2013 da pag. 21
 Lettori: 1.179.000 Diffusione: 266.088 Dir. Resp.: Roberto Napoletano da pag. 21 Lettori: 1.179.000 Diffusione: 266.088 Dir. Resp.: Roberto Napoletano da pag. 4 Lettori: 318.000 Diffusione: 98.223 Dir.
Lettori: 1.179.000 Diffusione: 266.088 Dir. Resp.: Roberto Napoletano da pag. 21 Lettori: 1.179.000 Diffusione: 266.088 Dir. Resp.: Roberto Napoletano da pag. 4 Lettori: 318.000 Diffusione: 98.223 Dir.
Uso dei biosimilari e sostenibilità: il punto di vista delle aziende farmaceutiche
 Uso dei biosimilari e sostenibilità: il punto di vista delle aziende farmaceutiche Monica Mecchia, Gruppo Biotecnologie di Farmindustria 24 ottobre 2018 Che cos è un farmaco biosimilare Il biosimilare
Uso dei biosimilari e sostenibilità: il punto di vista delle aziende farmaceutiche Monica Mecchia, Gruppo Biotecnologie di Farmindustria 24 ottobre 2018 Che cos è un farmaco biosimilare Il biosimilare
Position Paper sui farmaci biosimilari della Società Italiana di Farmacia Ospedaliera e dei Servizi Farmaceutici delle Aziende Sanitarie (SIFO)
") Position Paper sui farmaci biosimilari della Società Italiana di Farmacia Ospedaliera e dei Servizi Farmaceutici delle Aziende Sanitarie (SIFO) SIFO Società Italiana di Farmacia Ospedaliera e dei Servizi
Position Paper sui farmaci biosimilari della Società Italiana di Farmacia Ospedaliera e dei Servizi Farmaceutici delle Aziende Sanitarie (SIFO) SIFO Società Italiana di Farmacia Ospedaliera e dei Servizi
Farmaci di sintesi tradizionale Farmaci Biotecnologici Generici e Biosimilari
 Farmaci di sintesi tradizionale Farmaci Biotecnologici Generici e Biosimilari Farmaco di sintesi tradizionale ( small chemical entity ): definizione e caratteristiche È generalmente una molecola di piccole
Farmaci di sintesi tradizionale Farmaci Biotecnologici Generici e Biosimilari Farmaco di sintesi tradizionale ( small chemical entity ): definizione e caratteristiche È generalmente una molecola di piccole
Il farmaco generico, ormai ridefinito farmaco equivalente, è considerato essenzialmente simile al medicinale già in commercio (farmaco originatore)
") 1 Il farmaco generico, ormai ridefinito farmaco equivalente, è considerato essenzialmente simile al medicinale già in commercio (farmaco originatore) dal punto di vista qualitativo e quantitativo ma con
1 Il farmaco generico, ormai ridefinito farmaco equivalente, è considerato essenzialmente simile al medicinale già in commercio (farmaco originatore) dal punto di vista qualitativo e quantitativo ma con
I BIOSIMILARI LA POSIZIONE DELLE AZIENDE FARMACEUTICHE
 I BIOSIMILARI LA POSIZIONE DELLE AZIENDE FARMACEUTICHE EXECUTIVE SUMMARY Con oltre 7.000 farmaci in fase avanzata di sviluppo per diverse malattie, l innovazione farmacologica offrirà a breve molte nuove
I BIOSIMILARI LA POSIZIONE DELLE AZIENDE FARMACEUTICHE EXECUTIVE SUMMARY Con oltre 7.000 farmaci in fase avanzata di sviluppo per diverse malattie, l innovazione farmacologica offrirà a breve molte nuove
Farmaci bioequivalenti o generici: ostacolo o opportunità per l innovazione terapeutica?
 Farmaci bioequivalenti o generici: ostacolo o opportunità per l innovazione terapeutica? Roberto Trevisan USC Malattie Endocrine - Diabetologia ASST Papa Giovanni XXIII (Bergamo) Background I farmaci costano.
Farmaci bioequivalenti o generici: ostacolo o opportunità per l innovazione terapeutica? Roberto Trevisan USC Malattie Endocrine - Diabetologia ASST Papa Giovanni XXIII (Bergamo) Background I farmaci costano.
24/10/2018 Secondo Position Paper AIFA: opinioni a confronto
 24/10/2018 Secondo Position Paper AIFA: opinioni a confronto Manlio Florenzano, Coordinatore IBG Il nostro identikit: 12 anni di pratica clinica e oltre 20 prodotti in commercio in Italia PAZIENTI Ogni
24/10/2018 Secondo Position Paper AIFA: opinioni a confronto Manlio Florenzano, Coordinatore IBG Il nostro identikit: 12 anni di pratica clinica e oltre 20 prodotti in commercio in Italia PAZIENTI Ogni
ISTITUTO SUPERIORE DI SANITA'
 ISTITUTO SUPERIORE DI SANITA' DECRETO 26 aprile 2002 (Gazzetta Ufficiale serie generale del 7 maggio 2002, pagine 68-73) Accertamento della composizione e innocuità dei farmaci di nuova istituzione prima
ISTITUTO SUPERIORE DI SANITA' DECRETO 26 aprile 2002 (Gazzetta Ufficiale serie generale del 7 maggio 2002, pagine 68-73) Accertamento della composizione e innocuità dei farmaci di nuova istituzione prima
DISEGNO DI LEGGE. Senato della Repubblica XVI LEGISLATURA N. 1875
 Senato della Repubblica XVI LEGISLATURA N. 1875 DISEGNO DI LEGGE d iniziativa dei senatori CURSI e TOMASSINI COMUNICATO ALLA PRESIDENZA L 11 NOVEMBRE 2009 Modifiche all articolo 7 del decreto-legge 18
Senato della Repubblica XVI LEGISLATURA N. 1875 DISEGNO DI LEGGE d iniziativa dei senatori CURSI e TOMASSINI COMUNICATO ALLA PRESIDENZA L 11 NOVEMBRE 2009 Modifiche all articolo 7 del decreto-legge 18
È opportuno introdurre una chiara indicazione sull etichettatura del farmaco per assicurare un informazione trasparente sul medicinale utilizzato.
 I biosimilari La posizione delle Aziende farmaceutiche Executive summary I farmaci biosimilari usati in modo appropriato possono essere uno strumento per liberare risorse da allocare per un rapido e universale
I biosimilari La posizione delle Aziende farmaceutiche Executive summary I farmaci biosimilari usati in modo appropriato possono essere uno strumento per liberare risorse da allocare per un rapido e universale
GENERICI E BIOSIMILARI A CONFRONTO: PROBLEMATICHE FARMACOLOGICHE E REGOLATORIE
 XXXI CONGRESSO NAZIONALE SIFO CAGLIARI 6-8 OTTOBRE 2010 GENERICI E BIOSIMILARI A CONFRONTO: PROBLEMATICHE FARMACOLOGICHE E REGOLATORIE DOTT.ssa ENRICA MARIA PROLI DOTT.ssa ELENA JACOBONI COSA SONO I FARMACI
XXXI CONGRESSO NAZIONALE SIFO CAGLIARI 6-8 OTTOBRE 2010 GENERICI E BIOSIMILARI A CONFRONTO: PROBLEMATICHE FARMACOLOGICHE E REGOLATORIE DOTT.ssa ENRICA MARIA PROLI DOTT.ssa ELENA JACOBONI COSA SONO I FARMACI
L accesso alle cure oncologiche in Italia: come gestire tempi e differenze. Francesca Patarnello, VP Market Access & Government Affairs AstraZeneca
 L accesso alle cure oncologiche in Italia: come gestire tempi e differenze Francesca Patarnello, VP Market Access & Government Affairs AstraZeneca Ricerca, Innovazione e accesso: due mondi che non si parlano
L accesso alle cure oncologiche in Italia: come gestire tempi e differenze Francesca Patarnello, VP Market Access & Government Affairs AstraZeneca Ricerca, Innovazione e accesso: due mondi che non si parlano
Farmaci Innovativi. Appropriatezza: una corretta gestione delle risorse a supporto della ricerca. Qualità Efficacia Appropriatezza
 Farmaci Innovativi Qualità Efficacia Appropriatezza Garanzia dell universalità delle cure sul territorio nazionale TRA GRIFFATI, GENERICI E BIOSIMILARI: LA PAROLA D ORDINE E APPROPRIATEZZA Appropriatezza:
Farmaci Innovativi Qualità Efficacia Appropriatezza Garanzia dell universalità delle cure sul territorio nazionale TRA GRIFFATI, GENERICI E BIOSIMILARI: LA PAROLA D ORDINE E APPROPRIATEZZA Appropriatezza:
ENOXAPARINA SODICA E BIOSIMILARI
 ASL VCO - AZIENDA SANITARIA LOCALE VERBANO CUSIO OSSOLA ASL VC - AZIENDA SANITARIA LOCALE DI VERCELLI SOMMARIO SOMMARIO... 1 REDAZIONE DEL DOCUMENTO... 2 REFERENTE DOCUMENTO... 2 GRUPPO DI LAVORO INTERAZIENDALE...
ASL VCO - AZIENDA SANITARIA LOCALE VERBANO CUSIO OSSOLA ASL VC - AZIENDA SANITARIA LOCALE DI VERCELLI SOMMARIO SOMMARIO... 1 REDAZIONE DEL DOCUMENTO... 2 REFERENTE DOCUMENTO... 2 GRUPPO DI LAVORO INTERAZIENDALE...
AUDIZIONE Camera dei Deputati. Malattie rare. Pierluigi Russo. Roma, 29 Aprile 2015
 AUDIZIONE Camera dei Deputati Malattie rare Pierluigi Russo Roma, 29 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
AUDIZIONE Camera dei Deputati Malattie rare Pierluigi Russo Roma, 29 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
Studi Clinici in Pediatria e nelle Malattie Orfane
 Studi Clinici in Pediatria e nelle Malattie Orfane Valeria Antenucci OPBG Clinical & Research Services Verona, 19 Giugno 2014 Sperimentazione Clinica La sperimentazione farmacologica sull'uomo impone sempre
Studi Clinici in Pediatria e nelle Malattie Orfane Valeria Antenucci OPBG Clinical & Research Services Verona, 19 Giugno 2014 Sperimentazione Clinica La sperimentazione farmacologica sull'uomo impone sempre
Il Farmaco Veterinario: l utilizzo off-label e i generici
 Audizione Commissione Senato Roma, 14 marzo 2017 Il Farmaco Veterinario: l utilizzo off-label e i generici M. Fornasier Medico Veterinario - SIVAL Disegno di legge n. 540 e 499: che cosa deve fare il veterinario?
Audizione Commissione Senato Roma, 14 marzo 2017 Il Farmaco Veterinario: l utilizzo off-label e i generici M. Fornasier Medico Veterinario - SIVAL Disegno di legge n. 540 e 499: che cosa deve fare il veterinario?
MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE
 1+99 /2014 l CLASSIFICAZIONE Al SENSI DELL'ART.12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli 8 e 9
1+99 /2014 l CLASSIFICAZIONE Al SENSI DELL'ART.12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli 8 e 9
Sinergie e opportunità per l'innovazione e la competitività: il nuovo ruolo di AIFA
 Sinergie e opportunità per l'innovazione e la competitività: il nuovo ruolo di AIFA Prof. Sergio Pecorelli Presidente AIFA Lecce, 5 Dicembre 2014 S.Pecorelli@aifa.gov.it Il contesto globale Il nuovo contesto
Sinergie e opportunità per l'innovazione e la competitività: il nuovo ruolo di AIFA Prof. Sergio Pecorelli Presidente AIFA Lecce, 5 Dicembre 2014 S.Pecorelli@aifa.gov.it Il contesto globale Il nuovo contesto
Ricerca e farmaci antitumorali
 Ricerca e farmaci antitumorali Ad oggi disponibili 132 farmaci antitumorali Negli ultimi 15 anni immessi sul mercato 63. Circa 1 sostanza su 10 mila supera le prove per essere approvata Non più di 2 farmaci
Ricerca e farmaci antitumorali Ad oggi disponibili 132 farmaci antitumorali Negli ultimi 15 anni immessi sul mercato 63. Circa 1 sostanza su 10 mila supera le prove per essere approvata Non più di 2 farmaci
La nuova legislazione sulla Farmacovigilanza. Il sistema di qualità di farmacovigilanza in AIFA. Pietro Erba. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza Il sistema di qualità di farmacovigilanza in AIFA Pietro Erba Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa
La nuova legislazione sulla Farmacovigilanza Il sistema di qualità di farmacovigilanza in AIFA Pietro Erba Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa
AIFA Agenzia Italiana Del Farmaco Becaplermina (Regranex)
") AIFA - Becaplermina (Regranex) 19/02/2010 (Livello 2) AIFA Agenzia Italiana Del Farmaco Becaplermina (Regranex) Comunicato Stampa EMEA Domande e Risposte (FAQ) file:///c /documenti/reg1571.htm [19/02/2010
AIFA - Becaplermina (Regranex) 19/02/2010 (Livello 2) AIFA Agenzia Italiana Del Farmaco Becaplermina (Regranex) Comunicato Stampa EMEA Domande e Risposte (FAQ) file:///c /documenti/reg1571.htm [19/02/2010
Genova - 9 febbraio 2013
 FARMACO DI MARCA O EQUIVALENTE? Qualità, efficacia e responsabilità nell'attività prescrittiva Genova - 9 febbraio 2013 Il tema della Sostituibilità alla luce delle più recenti normative legislative. Walter
FARMACO DI MARCA O EQUIVALENTE? Qualità, efficacia e responsabilità nell'attività prescrittiva Genova - 9 febbraio 2013 Il tema della Sostituibilità alla luce delle più recenti normative legislative. Walter
Gestire l innovazione Terapeutica: Registri di monitoraggio AIFA e Farmacovigilanza
 Gestire l innovazione Terapeutica: Registri di monitoraggio AIFA e Farmacovigilanza Stefania Giorgetti UOC Farmacia Ospedaliera ASUR Marche Area Vasta 3 Macerata NUOVO NUOVA ENTITA CHIMICA INNOVATIVO INNOVATIVITA
Gestire l innovazione Terapeutica: Registri di monitoraggio AIFA e Farmacovigilanza Stefania Giorgetti UOC Farmacia Ospedaliera ASUR Marche Area Vasta 3 Macerata NUOVO NUOVA ENTITA CHIMICA INNOVATIVO INNOVATIVITA
Documento di Posizione della Società Italiana di Reumatologia sulla. Prescrizione dei Farmaci Biosimilari
 delib Milano, 14 luglio 2015 Documento di Posizione della Società Italiana di Reumatologia sulla Prescrizione dei Farmaci Biosimilari La perdita della copertura brevettuale permette l entrata sulla scena
delib Milano, 14 luglio 2015 Documento di Posizione della Società Italiana di Reumatologia sulla Prescrizione dei Farmaci Biosimilari La perdita della copertura brevettuale permette l entrata sulla scena
La nuova legislazione sulla Farmacovigilanza
 La nuova legislazione sulla Farmacovigilanza Il Sistema di Gestione del Rischio Stefania De Santis Sigma-Tau IFR S.p.A., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma, 13 settembre 2012
La nuova legislazione sulla Farmacovigilanza Il Sistema di Gestione del Rischio Stefania De Santis Sigma-Tau IFR S.p.A., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma, 13 settembre 2012
Impatto dello switching dell Infliximab nell artrite reumatoide: la dimensione economica
 Impatto dello switching dell Infliximab nell artrite reumatoide: la dimensione economica Tuccori, M., Convertino, I., Ferraro, S., Leonardi, L., Lucenteforte, E.; Blandizzi, C.; Gini, R., Roberto, G. Lorenzoni,
Impatto dello switching dell Infliximab nell artrite reumatoide: la dimensione economica Tuccori, M., Convertino, I., Ferraro, S., Leonardi, L., Lucenteforte, E.; Blandizzi, C.; Gini, R., Roberto, G. Lorenzoni,
EMA ed esercizio di comparabilità: il metodologo. Michela Cinquini Istituto di Ricerche Farmacologiche Mario Negri IRCCS - Milano
 EMA ed esercizio di comparabilità: il metodologo Michela Cinquini Istituto di Ricerche Farmacologiche Mario Negri IRCCS - Milano EMA e definizione di BIOSIMILARE EMA. http://www.ema.europa.eu/../wc500176768.pdf
EMA ed esercizio di comparabilità: il metodologo Michela Cinquini Istituto di Ricerche Farmacologiche Mario Negri IRCCS - Milano EMA e definizione di BIOSIMILARE EMA. http://www.ema.europa.eu/../wc500176768.pdf
CRITERI PER LA VALUTAZIONE DELL INNOVATIVITA
 ALLEGATO 1 CRITERI PER LA VALUTAZIONE DELL INNOVATIVITA L'autorizzazione all'immissione in commercio di un medicinale per una specifica indicazione, la sua ammissione alla rimborsabilità e il possibile
ALLEGATO 1 CRITERI PER LA VALUTAZIONE DELL INNOVATIVITA L'autorizzazione all'immissione in commercio di un medicinale per una specifica indicazione, la sua ammissione alla rimborsabilità e il possibile
Scheda unica di segnalazione di sospetta reazione avversa
 DIPARTIMENTO ASSISTENZA FARMACEUTICA Scheda unica di segnalazione di sospetta reazione avversa Dott.ssa Elisa Iori Responsabile di farmacovigilanza Assicura la qualità dei dati delle schede Gestisce il
DIPARTIMENTO ASSISTENZA FARMACEUTICA Scheda unica di segnalazione di sospetta reazione avversa Dott.ssa Elisa Iori Responsabile di farmacovigilanza Assicura la qualità dei dati delle schede Gestisce il
Modelli Sanitari Europei a confronto tra innovazione tecnologica farmacologica e organizzativa
 Modelli Sanitari Europei a confronto tra innovazione tecnologica farmacologica e organizzativa Biosimilari: sostenibilità del sistema e personalizzazione delle terapie Milano 26 giugno 2015 Intervento
Modelli Sanitari Europei a confronto tra innovazione tecnologica farmacologica e organizzativa Biosimilari: sostenibilità del sistema e personalizzazione delle terapie Milano 26 giugno 2015 Intervento
Le potenzialità degli studi registrativi e post marketing
 Dr.ssa Silvia Adami Area Sanità e Sociale Settore Farmaceutico-Protesica -Dispositivi medici Rio Novo Dorsoduro, 3493 30123 -Venezia 1 Studio Clinico - Classificazioni per fasi degli studi clinici La classificazione
Dr.ssa Silvia Adami Area Sanità e Sociale Settore Farmaceutico-Protesica -Dispositivi medici Rio Novo Dorsoduro, 3493 30123 -Venezia 1 Studio Clinico - Classificazioni per fasi degli studi clinici La classificazione
ISTRUZIONE OPERATIVA PER LA REDAZIONE DEI REPORT DI VALUTAZIONE DEI DATI CLINICI
 Ist. 05 Pag. 1 di 6 ISTRUZIONE OPERATIVA PER LA REDAZIONE DEI REPORT DI VALUTAZIONE DEI DATI CLINICI 1. SCOPO... 2 2. APPLICABILITA... 2 3. RESPONSABILITA... 2 4. ATTIVITA... 2 4.1. Report Valutazione
Ist. 05 Pag. 1 di 6 ISTRUZIONE OPERATIVA PER LA REDAZIONE DEI REPORT DI VALUTAZIONE DEI DATI CLINICI 1. SCOPO... 2 2. APPLICABILITA... 2 3. RESPONSABILITA... 2 4. ATTIVITA... 2 4.1. Report Valutazione
EFFICACIA E SOSTENIBILITA DELLA TERAPIA INSULINICA CON BIOSIMILARI
 EFFICACIA E OTENIBILITA DELLA TERAPIA INULINICA CON BIOIMILARI C. Baggiore OC Diabetologia e Malatt. Metab. Che cos è un biosimilare? Il termine biosimilare rappresenta per lo più una designazione normativa
EFFICACIA E OTENIBILITA DELLA TERAPIA INULINICA CON BIOIMILARI C. Baggiore OC Diabetologia e Malatt. Metab. Che cos è un biosimilare? Il termine biosimilare rappresenta per lo più una designazione normativa
IL POSITION PAPER DI FEDERSANITA ANCI. PRINCIPALI PUNTI EMERGENTI E SPUNTI PER UN DIBATTITO
 Roma, 1 marzo 2017 Il valore dei farmaci biosimilari nella governance della spesa Lorenzo Terranova IL POSITION PAPER DI FEDERSANITA ANCI. PRINCIPALI PUNTI EMERGENTI E SPUNTI PER UN DIBATTITO sviluppo
Roma, 1 marzo 2017 Il valore dei farmaci biosimilari nella governance della spesa Lorenzo Terranova IL POSITION PAPER DI FEDERSANITA ANCI. PRINCIPALI PUNTI EMERGENTI E SPUNTI PER UN DIBATTITO sviluppo
Caratteristiche delle preparazioni farmaceutiche degli equivalenti: aspetti farmacologici e legislativi internazionali e nazionali. Francesca Mattioli
 FARMACO DI MARCA O EQUIVALENTE? Qualità, efficacia e responsabilità nell'attività prescrittiva Caratteristiche delle preparazioni farmaceutiche degli equivalenti: aspetti farmacologici e legislativi internazionali
FARMACO DI MARCA O EQUIVALENTE? Qualità, efficacia e responsabilità nell'attività prescrittiva Caratteristiche delle preparazioni farmaceutiche degli equivalenti: aspetti farmacologici e legislativi internazionali
Legislazione sui farmaci equivalenti, provvedimenti regionali, risparmi. farmaci genericati. Dott.ssa Maria Susanna Rivetti
 Legislazione sui farmaci equivalenti, provvedimenti regionali, risparmi realizzati e possibili con l uso dei farmaci genericati Dott.ssa Maria Susanna Rivetti NORMATIVA DI RIFERIMENTO LA LEGGE 425/1996
Legislazione sui farmaci equivalenti, provvedimenti regionali, risparmi realizzati e possibili con l uso dei farmaci genericati Dott.ssa Maria Susanna Rivetti NORMATIVA DI RIFERIMENTO LA LEGGE 425/1996
Michela Franzin. Servizio di Farmacia. IRCCS Ospedale San Raffaele
 Michela Franzin Servizio di Farmacia IRCCS Ospedale San Raffaele La quota di mercato dei farmaci biologici con brevetto in scadenza tra il 2015 ed il 2020 è rilevante Potenziale risparmio derivante dalla
Michela Franzin Servizio di Farmacia IRCCS Ospedale San Raffaele La quota di mercato dei farmaci biologici con brevetto in scadenza tra il 2015 ed il 2020 è rilevante Potenziale risparmio derivante dalla
La nuova legislazione sulla Farmacovigilanza
 La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell Azienda Farmaceutica Alessandra Del Porto MSD Italia S.r.l., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma,
La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell Azienda Farmaceutica Alessandra Del Porto MSD Italia S.r.l., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma,
CRITERI PER LA VALUTAZIONE DEI FARMACI C(nn) DA PARTE DELLA COMMISSIONE REGIONALE DEL FARMACO (CRF)
 DA PARTE DELLA COMMISSIONE REGIONALE DEL FARMACO (CRF)") Direzione Generale Cura della Persona, Salute e Welfare Commissione Regionale Farmaco (D.G.R. 1540/2006 e 392/2015) Documento PTR n. 211 relativo a: CRITERI PER LA VALUTAZIONE DEI FARMACI C(nn) DA PARTE
Direzione Generale Cura della Persona, Salute e Welfare Commissione Regionale Farmaco (D.G.R. 1540/2006 e 392/2015) Documento PTR n. 211 relativo a: CRITERI PER LA VALUTAZIONE DEI FARMACI C(nn) DA PARTE
Dinamiche di erogazione, qualità dei servizi e contenimento dei costi: Il ruolo del Farmacista. Dott. Adriano Cristinziano
 Dinamiche di erogazione, qualità dei servizi e contenimento dei costi: Il ruolo del Farmacista Dott. Adriano Cristinziano Premessa Centralità del farmaco nei processi di cura. La maggior parte dei percorsi
Dinamiche di erogazione, qualità dei servizi e contenimento dei costi: Il ruolo del Farmacista Dott. Adriano Cristinziano Premessa Centralità del farmaco nei processi di cura. La maggior parte dei percorsi
Bioequivalenti e biosimilari: aspetti regolatori e loro impatto sull appropriatezza prescrittiva
 Bioequivalenti e biosimilari: aspetti regolatori e loro impatto sull appropriatezza prescrittiva Pierluigi Navarra Istituto di Farmacologia Facoltà di Medicina e Chirurgia Università Cattolica - Roma Analisi
Bioequivalenti e biosimilari: aspetti regolatori e loro impatto sull appropriatezza prescrittiva Pierluigi Navarra Istituto di Farmacologia Facoltà di Medicina e Chirurgia Università Cattolica - Roma Analisi
PROBLEMATICHE E NORMATIVE LEGATE ALLA MESSA A PUNTO DI UN VACCINO. Loredana Locatelli XIII Convegno Nazionale S.I.P.I. Abano Terme, 27 ottobre 2006
 PROBLEMATICHE E NORMATIVE LEGATE ALLA MESSA A PUNTO DI UN VACCINO Loredana Locatelli XIII Convegno Nazionale S.I.P.I. Abano Terme, 27 ottobre 2006 Agenda Definizioni Normative comunitarie e nazionali Procedure
PROBLEMATICHE E NORMATIVE LEGATE ALLA MESSA A PUNTO DI UN VACCINO Loredana Locatelli XIII Convegno Nazionale S.I.P.I. Abano Terme, 27 ottobre 2006 Agenda Definizioni Normative comunitarie e nazionali Procedure
La nuova legislazione sulla Farmacovigilanza. Trasparenza e comunicazione nella farmacovigilanza. Carmela Macchiarulo. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza Trasparenza e comunicazione nella farmacovigilanza Carmela Macchiarulo Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse
La nuova legislazione sulla Farmacovigilanza Trasparenza e comunicazione nella farmacovigilanza Carmela Macchiarulo Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse
Allegato A Prescrizione, impiego ed erogazione di farmaci off-label
 Allegato A Prescrizione, impiego ed erogazione di farmaci off-label PREMESSA L'autorizzazione all'immissione in commercio (AIC) per un medicinale viene rilasciata, dagli enti regolatori preposti, a seguito
Allegato A Prescrizione, impiego ed erogazione di farmaci off-label PREMESSA L'autorizzazione all'immissione in commercio (AIC) per un medicinale viene rilasciata, dagli enti regolatori preposti, a seguito
Commissione Regionale Farmaco MEDICINALI BIOSIMILARI DELL ERITROPOIETINA
 Commissione Regionale Farmaco (D.G.R. 1540/2006 e D.G.R. 2330/2008) Documento PTR n.119 relativo a: MEDICINALI BIOSIMILARI DELL ERITROPOIETINA Novembre 2010 Premessa...3 Confronto delle indicazioni terapeutiche
Commissione Regionale Farmaco (D.G.R. 1540/2006 e D.G.R. 2330/2008) Documento PTR n.119 relativo a: MEDICINALI BIOSIMILARI DELL ERITROPOIETINA Novembre 2010 Premessa...3 Confronto delle indicazioni terapeutiche
1. Medicinali di terapia cellulare somatica e medicinali di ingegneria tissutale
 Allegato 1 1. Medicinali di terapia cellulare somatica e medicinali di ingegneria tissutale La sostanza attiva del medicinale è rappresentata da cellule e/o tessuti prodotti dall ingegneria cellulare o
Allegato 1 1. Medicinali di terapia cellulare somatica e medicinali di ingegneria tissutale La sostanza attiva del medicinale è rappresentata da cellule e/o tessuti prodotti dall ingegneria cellulare o
Farmacovigilanza. Obbligo segnalazioni avverse entro 48h. Il Decreto in Gazzetta Ufficiale
 Farmacovigilanza. Obbligo segnalazioni avverse entro 48h. Il Decreto in Gazzetta Ufficiale Il provvedimento recepisce le direttive europee sulla farmacovigilanza. Tempi ridotti a 36 ore per segnalazione
Farmacovigilanza. Obbligo segnalazioni avverse entro 48h. Il Decreto in Gazzetta Ufficiale Il provvedimento recepisce le direttive europee sulla farmacovigilanza. Tempi ridotti a 36 ore per segnalazione
d iniziativa dei senatori RIZZOTTI, PICHETTO FRATIN, PEROSINO, BERUTTI, MASINI e FLORIS
 Senato della Repubblica LEGISLATURA N. 177 DISEGNO DI LEGGE d iniziativa dei senatori RIZZOTTI, PICHETTO FRATIN, PEROSINO, BERUTTI, MASINI e FLORIS COMUNICATO ALLA PRESIDENZA IL 28 MARZO 2018 Modifiche
Senato della Repubblica LEGISLATURA N. 177 DISEGNO DI LEGGE d iniziativa dei senatori RIZZOTTI, PICHETTO FRATIN, PEROSINO, BERUTTI, MASINI e FLORIS COMUNICATO ALLA PRESIDENZA IL 28 MARZO 2018 Modifiche
FARMACI: EMA, NEL 2012 RICEVUTE 96 RICHIESTE AIC, 59 PARERI POSITIVI CHMP = IL BILANCIO ANNUALE DELL' ATTIVITA' DELL' AGENZIA REGOLATORIA UE
 FARMACI: EMA, NEL 2012 RICEVUTE 96 RICHIESTE AIC, 59 PARERI POSITIVI CHMP = IL BILANCIO ANNUALE DELL' ATTIVITA' DELL' AGENZIA REGOLATORIA UE Roma, 17 apr. (Adnkronos Salute) - L' Agenzia europea dei medicinali
FARMACI: EMA, NEL 2012 RICEVUTE 96 RICHIESTE AIC, 59 PARERI POSITIVI CHMP = IL BILANCIO ANNUALE DELL' ATTIVITA' DELL' AGENZIA REGOLATORIA UE Roma, 17 apr. (Adnkronos Salute) - L' Agenzia europea dei medicinali
Definizione prezzo farmaci e politica del pricing
 1 Aspetti regolatori La determinazione del prezzo dei farmaci rimborsati dal SSN, avviene mediante la contrattazione tra Agenzia Italiana del Farmaco e le Aziende Farmaceutiche (ai sensi della Legge 326/2003
1 Aspetti regolatori La determinazione del prezzo dei farmaci rimborsati dal SSN, avviene mediante la contrattazione tra Agenzia Italiana del Farmaco e le Aziende Farmaceutiche (ai sensi della Legge 326/2003
PROGETTO DI FARMACOVIGILANZA ATTIVA
 Centro Regionale di Documentazione e Informazione sul Farmaco PROGETTO DI FARMACOVIGILANZA ATTIVA Studio multiregionale di Farmacovigilanza attiva per valutazione di sicurezza ed appropriatezza prescrittiva
Centro Regionale di Documentazione e Informazione sul Farmaco PROGETTO DI FARMACOVIGILANZA ATTIVA Studio multiregionale di Farmacovigilanza attiva per valutazione di sicurezza ed appropriatezza prescrittiva
MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE
 UAE/AR/PFG ';?'65 /2013 CLASSIFICAZIONE Al SENSI DELL'ART. 12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli
UAE/AR/PFG ';?'65 /2013 CLASSIFICAZIONE Al SENSI DELL'ART. 12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli
Commissione Regionale Farmaco MEDICINALI BIOSIMILARI DEL FILGRASTIM
 Commissione Regionale Farmaco (D.G.R. 1540/2006 e D.G.R. 2330/2008) Documento PTR n. 117 relativo a: MEDICINALI BIOSIMILARI DEL FILGRASTIM Ottobre 2010 Premessa...3 Confronto delle indicazioni terapeutiche
Commissione Regionale Farmaco (D.G.R. 1540/2006 e D.G.R. 2330/2008) Documento PTR n. 117 relativo a: MEDICINALI BIOSIMILARI DEL FILGRASTIM Ottobre 2010 Premessa...3 Confronto delle indicazioni terapeutiche
AGENZIA ITALIANA DEL FARMACO
 AGENZIA ITALIANA DEL FARMACO DETERMINA 12 novembre 2014 Regime di rimborsabilita' e prezzo del medicinale per uso umano «Sovaldi» (sofosbuvir), autorizzata con procedura centralizzata europea dalla Commissione
AGENZIA ITALIANA DEL FARMACO DETERMINA 12 novembre 2014 Regime di rimborsabilita' e prezzo del medicinale per uso umano «Sovaldi» (sofosbuvir), autorizzata con procedura centralizzata europea dalla Commissione
ALTERNATIVA CHE FA LA DIFFERENZA
 n.1 Equivalenti e Uguali Zentiva Oltre i farmaci di marca: UN MONDO DI VANTAGGI IN EVIDENZA Efficacia terapeutica e risparmio: SI PUÒ! Non solo equivalenti. Ecco GLI UGUALI! Come scegliere un farmaco?
n.1 Equivalenti e Uguali Zentiva Oltre i farmaci di marca: UN MONDO DI VANTAGGI IN EVIDENZA Efficacia terapeutica e risparmio: SI PUÒ! Non solo equivalenti. Ecco GLI UGUALI! Come scegliere un farmaco?
Centro Ricerca Interdipartimentale Farmacogenetica Farmacogenomica
 Armando A. Genazzani Università del Piemonte Orientale genazzani@pharm.unipmn.it 347.9829981 Centro Ricerca Interdipartimentale Farmacogenetica Farmacogenomica Principi generali del comparability exercise
Armando A. Genazzani Università del Piemonte Orientale genazzani@pharm.unipmn.it 347.9829981 Centro Ricerca Interdipartimentale Farmacogenetica Farmacogenomica Principi generali del comparability exercise
L industria: oltre il farmaco e la ricerca
 L industria: oltre il farmaco e la ricerca Dott. Giorgio Ghignoni Direttore Esecutivo Public Affairs & Policy Pfizer Italia Convegno MALATTIE RARE E DISABILITA Palazzo Senatorio Aula Giulio Cesare Roma,
L industria: oltre il farmaco e la ricerca Dott. Giorgio Ghignoni Direttore Esecutivo Public Affairs & Policy Pfizer Italia Convegno MALATTIE RARE E DISABILITA Palazzo Senatorio Aula Giulio Cesare Roma,
DECRETI E DELIBERE DI ALTRE AUTORITÀ
 DECRETI E DELIBERE DI ALTRE AUTORITÀ DETERMINAZIONE 7 luglio 2010. AGENZIA ITALIANA DEL FARMACO Regime di rimborsabilità e prezzo di vendita del medicinale «Victoza». (Determinazione/C 397/2010). Regime
DECRETI E DELIBERE DI ALTRE AUTORITÀ DETERMINAZIONE 7 luglio 2010. AGENZIA ITALIANA DEL FARMACO Regime di rimborsabilità e prezzo di vendita del medicinale «Victoza». (Determinazione/C 397/2010). Regime
Oggetto: Direttive alle Aziende Sanitarie relative alla gestione dei Registri di monitoraggio dell Agenzia Italiana del Farmaco (AIFA).
.") Oggetto: Direttive alle Aziende Sanitarie relative alla gestione dei Registri di monitoraggio dell Agenzia Italiana del Farmaco (AIFA). L Assessore dell Igiene e Sanità e dell Assistenza Sociale riferisce
Oggetto: Direttive alle Aziende Sanitarie relative alla gestione dei Registri di monitoraggio dell Agenzia Italiana del Farmaco (AIFA). L Assessore dell Igiene e Sanità e dell Assistenza Sociale riferisce
REGOLAMENTO (UE) / DELLA COMMISSIONE. del
 / DELLA COMMISSIONE. del") COMMISSIONE EUROPEA Bruxelles, 29.5.2018 C(2018) 3193 final REGOLAMENTO (UE) / DELLA COMMISSIONE del 29.5.2018 che modifica il regolamento (CE) n. 847/2000 per quanto riguarda la definizione del concetto
COMMISSIONE EUROPEA Bruxelles, 29.5.2018 C(2018) 3193 final REGOLAMENTO (UE) / DELLA COMMISSIONE del 29.5.2018 che modifica il regolamento (CE) n. 847/2000 per quanto riguarda la definizione del concetto
L aggiornamento pubblicato nel marzo del 2007 è stato integrato con la pubblicazione della prima lista di farmaci essenziali ad uso pediatrico
 Nel 1977, l Organizzazione Mondiale della Sanità (OMS) pubblica il primo report sui farmaci essenziali. Indicato con l acronimo WHO TRS 615, il report tecnico costituisce la prima lista di farmaci essenziali
Nel 1977, l Organizzazione Mondiale della Sanità (OMS) pubblica il primo report sui farmaci essenziali. Indicato con l acronimo WHO TRS 615, il report tecnico costituisce la prima lista di farmaci essenziali
È un medicinale che contiene lo stesso principio attivo e nella stessa concentrazione di un farmaco di marca non più coperto da brevetto(definito
 CHE COS'È UN FARMACO EQUIVALENTE? È un medicinale che contiene lo stesso principio attivo e nella stessa concentrazione di un farmaco di marca non più coperto da brevetto(definito originator). I farmaci
CHE COS'È UN FARMACO EQUIVALENTE? È un medicinale che contiene lo stesso principio attivo e nella stessa concentrazione di un farmaco di marca non più coperto da brevetto(definito originator). I farmaci
Medicinali Biosimilari
 Commissione Europea Cosa c è da sapere riguardo ai Medicinali Biosimilari Informazioni per i pazienti Un documento di consenso CRESCITA Questa informativa condivisa sui medicinali biosimilari è stata redatta
Commissione Europea Cosa c è da sapere riguardo ai Medicinali Biosimilari Informazioni per i pazienti Un documento di consenso CRESCITA Questa informativa condivisa sui medicinali biosimilari è stata redatta
Diabete e biosimilari dell insulina: evidenze cliniche
 Diabete e biosimilari dell insulina: evidenze cliniche Francesco Rossi I progressi in farmacologia: le biotecnologie 1 La crescita dell industria biotech in Italia Anche in Italia, l industria biotecnologica
Diabete e biosimilari dell insulina: evidenze cliniche Francesco Rossi I progressi in farmacologia: le biotecnologie 1 La crescita dell industria biotech in Italia Anche in Italia, l industria biotecnologica
DETERMINAZIONE 12 luglio Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010).
.") DETERMINAZIONE 12 luglio 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010). Regime di rimborsabilità e prezzo di vendita della specialità medicinale
DETERMINAZIONE 12 luglio 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010). Regime di rimborsabilità e prezzo di vendita della specialità medicinale
Obiettivi delle Reti Oncologiche Regionali
 Obiettivi delle Reti Oncologiche Regionali Garanzia della qualità/standard assistenziali Appropriatezza diagnostica e terapeutica Razionalizzazione dei servizi Integrazione dei PDTA Razionalizzazione delle
Obiettivi delle Reti Oncologiche Regionali Garanzia della qualità/standard assistenziali Appropriatezza diagnostica e terapeutica Razionalizzazione dei servizi Integrazione dei PDTA Razionalizzazione delle
Farmaci originator, biosimilari e Antitrust
 I biosimilari, il rispetto delle regole e la competition Francesco Mazza Direttore della Direzione Legale, Fiscale e Compliance Milano, 30 maggio 2017 Il valore industriale della farmaceutica in Italia
I biosimilari, il rispetto delle regole e la competition Francesco Mazza Direttore della Direzione Legale, Fiscale e Compliance Milano, 30 maggio 2017 Il valore industriale della farmaceutica in Italia
Art.1 (Oggetto del decreto legislativo)
") Schema di decreto legislativo di modifica al decreto legislativo 24 aprile 2006, n. 219 recante Attuazione della direttiva 2001/83/CE (e successive direttive di modifica) relativa ad un codice comunitario
Schema di decreto legislativo di modifica al decreto legislativo 24 aprile 2006, n. 219 recante Attuazione della direttiva 2001/83/CE (e successive direttive di modifica) relativa ad un codice comunitario
AIFA Agenzia Italiana del Farmaco Benfluorex
 AIFA - Benfluorex 18/12/2009 (Livello 2) AIFA Agenzia Italiana del Farmaco Benfluorex Comunicato EMEA Domande e Risposte (FAQ) file:///c /documenti/ben950.htm [18/12/2009 15.50.04] 18 December 2009 EMA/CHM/815033/2009
AIFA - Benfluorex 18/12/2009 (Livello 2) AIFA Agenzia Italiana del Farmaco Benfluorex Comunicato EMEA Domande e Risposte (FAQ) file:///c /documenti/ben950.htm [18/12/2009 15.50.04] 18 December 2009 EMA/CHM/815033/2009
della ONDA SALUTE Informazione del paziente tra diritto e valore
 della ONDA SALUTE 2018 Informazione del paziente tra diritto e valore ONDA - CHI SIAMO Un Osservatorio che dal 2006 promuove la medicina di genere* a livello istituzionale, scientifico, sanitario-assistenziale
della ONDA SALUTE 2018 Informazione del paziente tra diritto e valore ONDA - CHI SIAMO Un Osservatorio che dal 2006 promuove la medicina di genere* a livello istituzionale, scientifico, sanitario-assistenziale
Presentazione: L uso dei biosimilari e la tutela del paziente
 Presentazione: L uso dei biosimilari e la tutela del paziente Relatore: Avv. Francesca Mastroianni 30 maggio 2017 Public Declaration of transparency/interests* Interests in pharmaceutical industry NO Current
Presentazione: L uso dei biosimilari e la tutela del paziente Relatore: Avv. Francesca Mastroianni 30 maggio 2017 Public Declaration of transparency/interests* Interests in pharmaceutical industry NO Current
Corso di aggiornamento in FARMACOVIGILANZA. Ospedale Evangelico Internazionale Genova, 12 e 17 Giugno 2013
 Corso di aggiornamento in FARMACOVIGILANZA Ospedale Evangelico Internazionale Genova, 12 e 17 Giugno 2013 Farmacovigilanza La scienza e le attività collegate alla identificazione, valutazione, conoscenza
Corso di aggiornamento in FARMACOVIGILANZA Ospedale Evangelico Internazionale Genova, 12 e 17 Giugno 2013 Farmacovigilanza La scienza e le attività collegate alla identificazione, valutazione, conoscenza