La distribuzione dei veri e falsi positivi la ricerca della giusta soglia
|
|
|
- Massimo Ferrara
- 5 anni fa
- Visualizzazioni
Transcript
1 La distribuzione dei veri e falsi positivi la ricerca della giusta soglia
2 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query in parole di pochi amminoacidi, di solito 2 o 3 (parametro W) e generando una lista di parole affini (diverso da FASTA) con la matrice di sostituzione (BLOSUM). Le parole affini conservate dovranno avere uno score superiore ad una soglia fissata T 2. Le parole affini sono ricercate nella banca dati per match esatti ed una volta trovate le sequenze che li contengono questi vengono estesi a dx e sx dell allineamento per una certa profondità stabilita dal parametro X e le coppie di segmenti, presenti nella stessa coppia di sequenze, che totalizzano un punteggio di similarità statisticamente significativo, superiore ad una soglia S, vengono definiti HSP (High scoring Segment Pairs). 3. Nella stessa coppia possono esserci più HSP di cui é anche possibile calcolare la probabilità di occorrenza (Karlin & Altschul, 1993). W = word-size T = threshold X = elongation S = HSP threshold
3 Si definisce MSP (Maximal scoring Segment Pair) la coppia di segmenti, di eguale lunghezza, che realizza il massimo punteggio di similarità nel confronto di due sequenze; l algorimo ne valuta in modo rigoroso la significatività statistica (Karlin & Altschul, 1990, 1993).
4 BLAST two hit method le versioni attuali di Blast adottano il Two-hit method che deriva dall'osservazione che il tempo di esecuzione dell'algoritmo e' principalmente impiegato nell'allungamento degli Hits per ottenere gli HSPs. L'algoritmo allora considera solo i casi in cui esistono due hit sulla stessa diagonale ad una distanza inferiore ad un parametro A prima di cercare gli HSPs. Per non perdere in sensibilità e' stata abbassata la soglia di T. L'algoritmo è più veloce e non ha perso in precisione Nella sua attuale implementazione, inoltre, BLAST considera anche i gap nel tentativo di unire, quindi, degli HSP "ungapped" che sono spazialmente vicini nella matrice di allineamento e la cui unione in un unico frammento (contenente gap ed inserzioni) non comporta un peggioramento dello score finale ma un miglioramento complessivo. Il tutto secondo dei nuovi parametri che regolano i costi e le penalità della presenza di gap nell'allineamento. Il parametro A
5 BLAST I vari algoritmi differiscono molto per il metodo con cui definiscono una sequenza casuale. BLAST calcola a priori la probabilità che un certo punteggio sia significativo sulla base della dimensione e composizione della banca dati applicando: λs E( S) = kmne dove m è la lunghezza della sequenza query e n è la lunghezza della sequenza subject della banca dati λ e K sono precalcolati secondo una distribuzione standard interna al contrario di FASTA. Il punteggio è simile a quello di FASTA La significatività di un risultato è espressa come valore E(S) (Expectation). Più basso il valore di E più significativo è l allineamento. Un valore di 1.0e-5 per esempio vuol dire che la probabilità di avere per caso una sequenza con lo stesso score della mia query è uguale a 1.0e-5; ovvero l attesa è che ogni sequenze se ne possa, in media, trovare una (1/100000) che totalizzi un punteggio uguale o migliore di 1.0e-5.

6
7
8 CONFRONTO BLAST E FASTA fasta3 proteina o DNA contro banca dati o proteici o DNA rispettivamente fastx/y3 DNA contro banca dati proteico, traduzione nei 6 frame tfastx/y3 proteina contro banca dtai di DNA tradotto blastn query DNA banca dati DNA blastp query proteina banca dati proteine blastx query DNA (tradotta nei sei frame di lettura) banca dati proteine tblastn query proteina banca dati DNA (sequenze tutte tradotte nelle sei fasi di lettura) tblastx query DNA (tradotta nei sei frame di lettura) banca dati DNA (tradotta nei sei frame di lettura) SIMILARITA USO DELLA SCORING MATRIX K-TUPLE FASTA Locale (e' in genere riportato solo il miglior allineamento locale) Durante la estensione nella fase B. In questo caso il calcolo si effettua nella prima fase solo per identità 1-2 aa / 4-6 nt BLAST Locale (e' riportata la serie di allineamenti locali sopra il valore soglia tra query e subject: al contrario di FASTA si riescono ad individuare repeat e zone eventuali di overlap tra gli allineamenti locali che sono eliminate da FASTA nella fase C) Fase di scansione per W e fase di estensione per gli HSP l'algoritmo e' ottimizzato per ricercare parole W "simili" e non esatte. Si traduce il tutto in una maggiore sensibilità di ricerca rispetto a FASTA per le proteine. GAP Consentiti nella fase C Consentiti nella versione attuale 3 aa / 11-12nt. A livello nucleotidico, non essendo applicate matrici di similarità che perdono di significato avendo solo 4 simboli (A,C,G,T), BLAST perde in sensibilità avendo W=11 VELOCITA' Da 1/2 ad 1/5 di BLAST Da 2 a 5 volte maggiore di FASTA SPECIFICITA' Migliore per il confronto di sequenze nucleotidiche Migliore per il confronto di sequenze proteiche
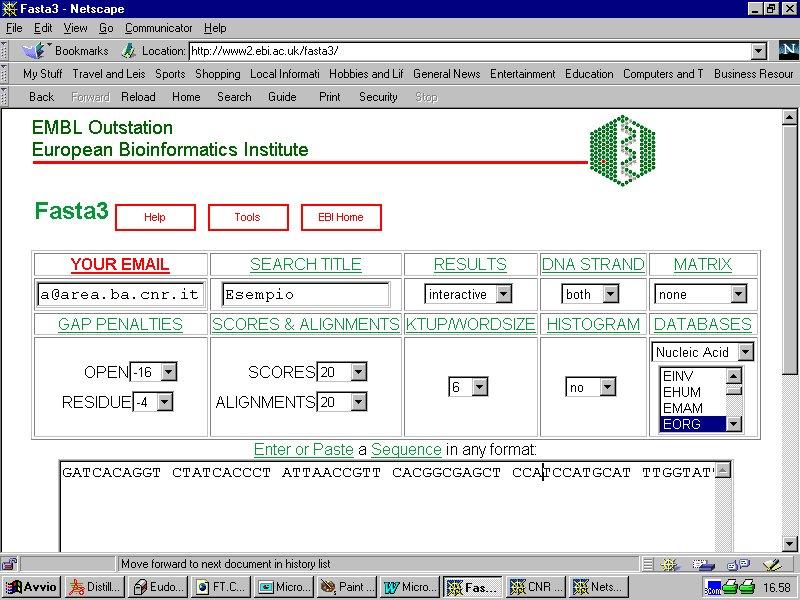
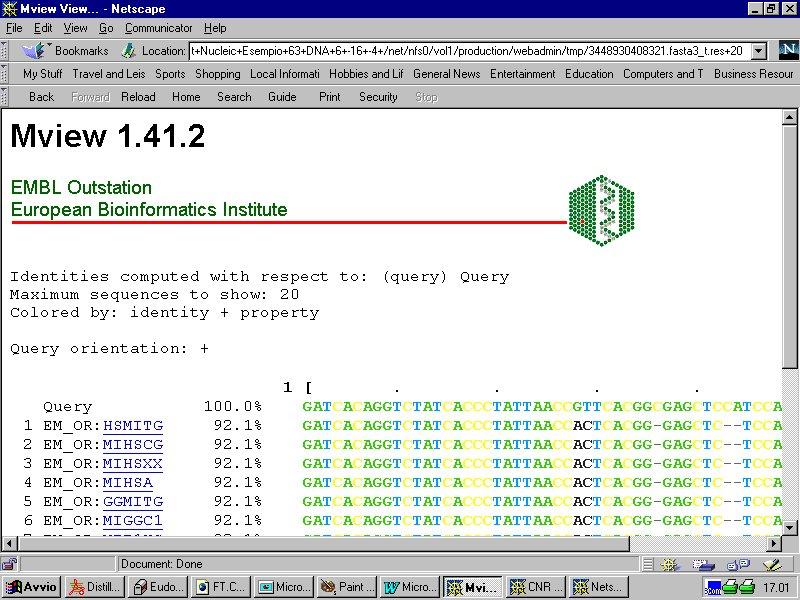
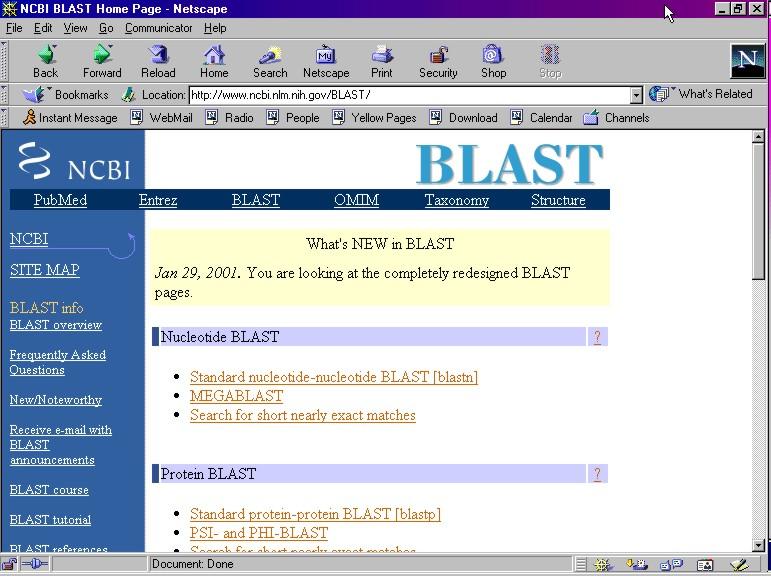
9 Alcuni esempi di interfacce web FASTA ( BLAST (
10
11

12
13
14
15

16

17
18

19
20
21 ALLINEAMENTI MULTIPLI Identificazione di siti funzionalmente importanti Dimostrazione di omologia Filogenesi molecolare Ricerca di somiglianze deboli ma significative in banche dati Predizione di struttura Predizione di funzione
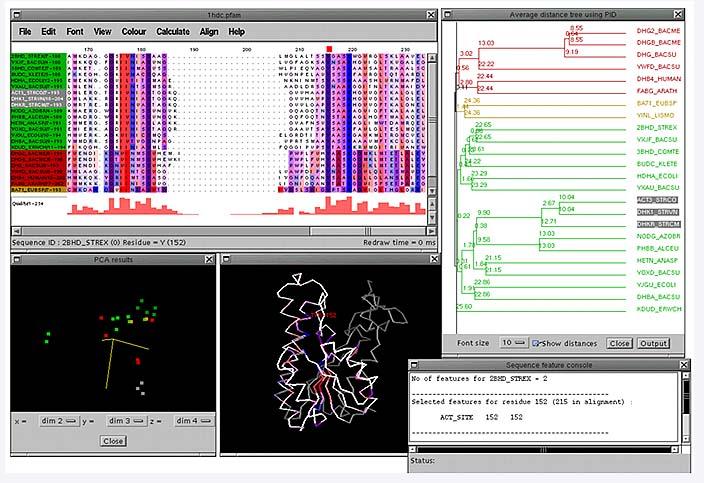
22

23 Utilizzo dei colori I file raw-text possono essere utilizzati per visualizzare le colonne, ma è possibile associare colori diversi per residui con caratteristiche chimico fisiche diverse. Questo facilita molto la visualizzazione dei multiallineamenti ESPript e PrettyPlot sono programmi dedicati a questo tipo di analisi qualitativa disponibili in rete
24 ESTENSIONE DEGLI ALLINEAMENTI GLOBALI (NW) O LOCALI (SW)? L applicazione degli algoritmi per la ricerca di un allineamento ottimale tra due sequenze pone problemi per l applicazione a più di tre sequenze contemporaneamente se L è la lunghezza delle sequenze occorrerebbe un tempo di O(L N ) che è impraticabile Uso di metodi approssimati (euristici) o progressivi che si basano sull ipotesi che le sequenze da allineare siano filogeneticamente correlate
25 Metodi approssimati Allineamento progressivo (Clustal) Metodi iterativi (Multalin) Metodi basati su zone comuni di sequenza conservate (Profili) Metodi statistici e modelli probabilistici (HMM)
26 Allineamento progressivo CLUSTAL (Higgins & Sharp, 1988) ClustalW ClustalX PILEUP (GCG)
27 CLUSTAL (Higgins & Sharp, 1988) 1. Allineamento a coppie di tutte le sequenze iniziali con: 1. Metodi approssimati (n-ple) oppure 2. algoritmo dinamico di Myers & Miller, Il punteggio degli allineamenti (matrice delle distanze) è utilizzato per costruire un albero filogenetico (neighbor-joining) 3. Allineamento delle sequenze secondo l ordine dell albero (le sequenze più simili prima)
28 neighbor-joining Saitou Mol. Biol. Evol È un algoritmo di clustering che attraverso iterazioni successive determina le coppie di sequenze più simili e le restanti. Se N sono le sequenze allora ci saranno N(N-1)/2 Possibilità di scegliere la prima coppia di sequenze che tra loro hanno il punteggio di similarità più alto. La prima coppia così costituita verrà utilizzata come consenso e la procedura si ripete per trovare un altra sequenza o cluster che sia il più vicino possibile alla coppia appena costituita. Parlando in termini filogenetici in cui NJ viene usato si può dire che l albero filogenetico si risolve progressivamente dalla tipologia a stella fino a che non si ottengono tutti gli N-3 rami interni.
29
30
31
32 In questo caso si ha che il nuovo nodo X, dato dall unione di (1-2), avrà una distanza dagli altri pari a: (m appartiene ai nodi {3,8}) D xm = 1 2 ( D + D D ) 1, m 2, m 1,2
33
34
35
36
37 CLUSTAL Il contributo delle sequenze al punteggio dell allineamento multiplo è pesato Sistema di penalizzazione degli indels che sono favoriti tra domini conservati. Durante il processo di allineamento, la penalizzazione dei gap viene abbassata nelle zone in cui sono già presenti dei gap Si basa sul NJ che utilizza i valori di similarità dei k(k-1)/2 allineamenti a coppie (basato sull idea dell algoritmo di Feng-Doolittle). Nella costruzione dell allineamento fa un allineamento sequenza -> profilo Sequence weighting: ogni sequenza ha un peso associato, funzione della distribuzione statistica delle sequenze. Gruppi di sequenze correlate hanno pesi diminuiti perchè contengono informazione ridondante. Matrix score: a seconda della distanza fra le sequenze sono usate diverse matrici di sostituzione. Special gap score: i punteggi associati ai gap variano in relazione a molti fattori, tra cui la frequenza dei residui allineati con il gap e la lunghezza delle sequenze.
38
39 QUALITA DI UN ALLINEAMENTO MULTIPLO WSP score = N i= 2 i 1 j= 1 W QUAL( A ij ij ) N CAGPHJKLCMMWERQASDF CAHPHJKLCVMWERQASDF CAGPHJELCVMWERRASDF MAGPHJKLCVMWERFASDF Si ottiene sommando i punteggi di similarità QUAL(A) pesati per un peso W di ciascuna delle possibili coppie allineate nell allineamento multiplo (Weight Sum of Pairs) Dipende dai parametri scelti per calcolare match e INDELS Il peso W serve per pesare sequenze sovra o sotto rappresentate nell allineamento
40
41 Svantaggi dei metodi progressivi Non c è garanzia di trovare la soluzione ottimale Gli errori iniziali sono propagati nei passaggi successivi. Se si introduce un errore nell allineamento iniziale non si può più correggere ma anzi si fissa Gli errori nell allineamento dipendono dalla somiglianza delle sequenze ovvero bisogna stare attenti alle sequenze in input che siano realmente omologhe e di lunghezza paragonabile tra loro per evitare inserzioni di troppi gap Gli alberi filogenetici iniziali derivano da matrici di distanza tra coppie di sequenze allineate separatamente che sono meno affidabili di alberi derivati da allineamenti multipli completi Quando le sequenze sono molto divergenti (25-30% di identità) i metodi progressivi sono poco affidabili
42 Metodi iterativi I metodi iterativi tentano di correggere errori iniziali riallineando iterativamente sottogruppi di sequenze che poi vengono riuniti in un allineamento multiplo MULTALIN (Corpet, 1988) PRRP (Gotoh, 1996)
43 Metodi iterativi Negli algoritmi precedenti, una volta che un allineamento è fissato, non viene più modificato nei passi successivi. In particolare, la posizione dei gap non cambia (once a gap, always a gap). In un metodo iterativo, una volta generato un allineamento iniziale, una sequenza o un insieme di sequenze è rimosso dall allineamento e riallineato al profilo relativo alle rimanenti sequenze. Si può dimostrare che, iterando su tutte le sequenze, si converge ad un massimo locale. Metodo di Barton-Sternberg Trova le due sequenze con il massimo grado di somiglianza e allineale con un algoritmo standard per il pairwise alignment. Trova la sequenza più simile al profilo del precedente allineamento e allineala a tale profilo. Ripeti finchè non sono state incluse tutte le sequenze. Rimuovi la prima sequenza e riallineala al profilo delle rimanenti. Ripeti per ogni sequenza. Ripeti il passo precedente finchè il punteggio non converge oppure fino a quando si raggiunge un numero massimo di iterazioni.
44 Punti fondamentali su allineamenti progressivi e iterativi 1) progressivi: Idea: costruire l allineamento multiplo aggiungendo una sequenza alla volta. Metodo euristico: non garantisce l ottimalità. Occorre stabilire: in quale ordine aggiungere le sequenze; come costruire la progressione; come allineare una sequenza ad un allineamento. La progressione può essere lineare aggiungi la sequenza all unico allineamento; oppure ad albero costruisci più sottoallineamenti e allineali in qualche modo tra loro Alberi guida le cui foglie sono sequenze e i cui nodi interni rappresentano gruppi (cluster ) di sequenze. Usati per determinare l ordine in cui effettuare l allineamento progressivo k(k-1)/2 confronti. definisci una distanza fra cluster. Ripeti i due passi seguenti fino ad ottenere un unico cluster: scegli i due cluster con distanza minima e fondili in un unico cluster; aggiorna le distanze calcolando la distanza tra il nuovo cluster e i rimanenti. Il procedimento genera un albero con radice. problema fondamentale è la propagazione dell errore che si può risolvere con i metodi iterativi 2) iterativi A B C D E riallineano iterativamente sottogruppi di sequenze che poi vengono riuniti in un allineamento multiplo
45 T-COFFEE (Notredame, JMB 2000) T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation) T-Coffee has two main features: It provides a simple and flexible means of generating multiple alignments, using heterogeneous data sources. 1. The data from these sources are provided to T-Coffee via a library of pair-wise alignments. T-Coffee computes multiple alignments using a library that was generated using a mixture of: local pair-wise alignments (lalign) global pair-wise alignments (clustalw) 2. The second main feature of T-Coffee is the optimization method, which is used to find the multiple alignment that best fits the pair-wise alignments in the input library. We use a so-called progressive strategy (Feng & Doolittle, 1987; Taylor, 1988; Thompson et al., 1994), which is similar to that used in ClustalW. This has the advantage of being fast and relatively robust. Use of a heuristic algorithm that called library extension. The overall idea is to combine information in such a manner that the final weight, for any pair of residues, reflects some of the information contained in the whole library. To do so, a triplet approach is used.
46
47 Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica a nessuna delle proteine del dataset. si possono definire dei simboli che la definiscano e che indichino anche conservazioni non perfette in una posizione. è possibile utilizzare una formattazione precisa che permetta di capire anche le variazioni in una posizione, non solo le conservazioni.

48 Alcuni modi di indicare le sequenze consenso WebLogo è una risorsa in rete per generare sequenze consenso Consenso esatto Consenso a simboli GRVQGV--R------A--LG -GWV GRVQGh-aRvvvvvvAvvLGivGWV GRVQG[VI]-[FY]R------A L----GWY GRVQGV--R-6A LG--GWV Consenso con variazioni Consenso con ripetizioni
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
ALLINEAMENTI MULTIPLI
 ALLINEAMENTI MULTIPLI Identificazione di siti funzionalmente importanti Dimostrazione di omologia Filogenesi molecolare Ricerca di somiglianze deboli ma significative in banche dati Predizione di struttura
ALLINEAMENTI MULTIPLI Identificazione di siti funzionalmente importanti Dimostrazione di omologia Filogenesi molecolare Ricerca di somiglianze deboli ma significative in banche dati Predizione di struttura
Z-score. lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random
/ deviazione standard random") Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Le sequenze consenso
 Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
La ricerca di similarità: i metodi
 La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
Allineamento multiplo
 Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Lezione 8. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 8 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Allineamento multiplo di sequenze
 Allineamento multiplo di sequenze Bioinformatica a.a. 2008/2009 Letterio Galletta Università di Pisa 22 Maggio 2009 Letterio Galletta (Università di Pisa) Allineamento multiplo di sequenze 22 Maggio 2009
Allineamento multiplo di sequenze Bioinformatica a.a. 2008/2009 Letterio Galletta Università di Pisa 22 Maggio 2009 Letterio Galletta (Università di Pisa) Allineamento multiplo di sequenze 22 Maggio 2009
SAGA: sequence alignment by genetic algorithm. ALESSANDRO PIETRELLI Soft Computing
 SAGA: sequence alignment by genetic algorithm ALESSANDRO PIETRELLI Soft Computing Bologna, 25 Maggio 2007 Multi Allineamento di Sequenze (MSAs) Cosa sono? A cosa servono? Come vengono calcolati Multi Allineamento
SAGA: sequence alignment by genetic algorithm ALESSANDRO PIETRELLI Soft Computing Bologna, 25 Maggio 2007 Multi Allineamento di Sequenze (MSAs) Cosa sono? A cosa servono? Come vengono calcolati Multi Allineamento
Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti Esercitazione 7 2.
 Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti patti@di.unito.it Esercitazione 7 1 Info&Bio Bio@Lab Allineamento di sequenze Esercitazione 7 2 1 Es2: Allineamento
Laboratorio di Informatica 2004/ 2005 Corso di laurea in biotecnologie - Novara Viviana Patti patti@di.unito.it Esercitazione 7 1 Info&Bio Bio@Lab Allineamento di sequenze Esercitazione 7 2 1 Es2: Allineamento
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Lezione 6. Ricerche in banche dati (databases) attraverso l uso di BLAST
 attraverso l uso di BLAST") Lezione 6 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Lezione 6 Ricerche in banche dati (databases) attraverso l uso di BLAST BLAST: Basic Local Alignment Search Tool Basic Local Alignment Search Tool. Altschul et al. 1990,1994,1997 Sviluppato per rendere
Allineamenti multipli
 Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Ricerca di omologia di sequenza
 Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
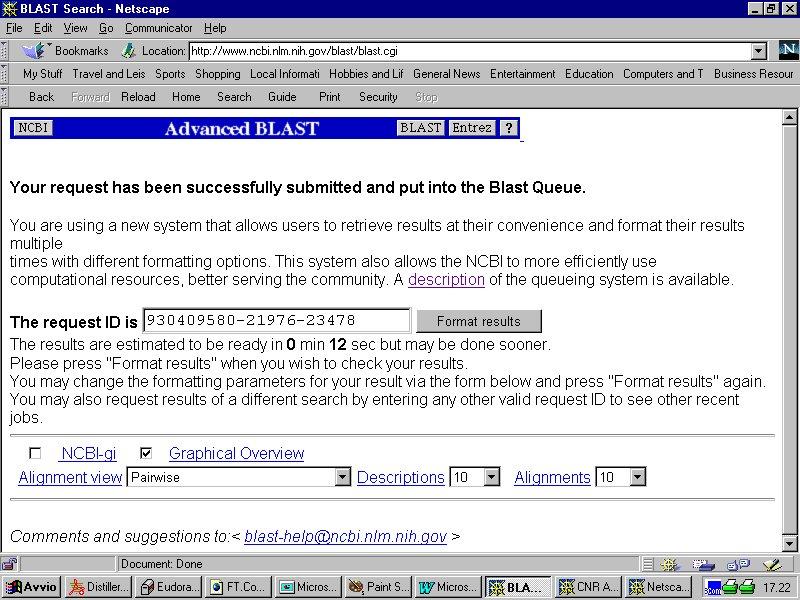
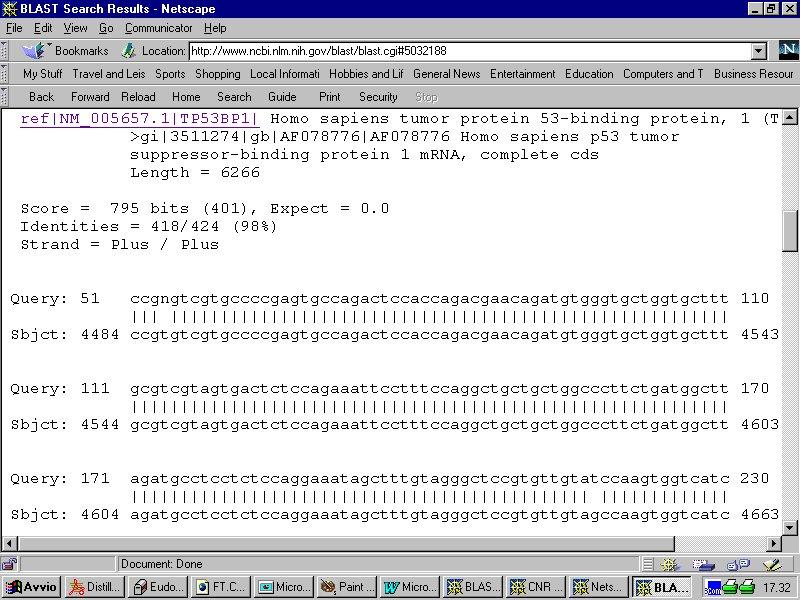
Esempio di utilizzo del programma BLAST disponibile all NCBI Form di Nucleotide BLAST
 Esempio di utilizzo del programma BLAST disponibile all NCBI www.ncbi.nlm.nih.gov/blast Form di Nucleotide BLAST Per un uso più avanzato, si possono impostare parametri particolari (es. cost to open gap,
Esempio di utilizzo del programma BLAST disponibile all NCBI www.ncbi.nlm.nih.gov/blast Form di Nucleotide BLAST Per un uso più avanzato, si possono impostare parametri particolari (es. cost to open gap,
Allineamenti Multipli di Sequenze
 Allineamenti Multipli di Sequenze 1 Allineamento multiplo di sequenze: obiettivi di oggi Definire un allineamento multiplo di sequenze; com è generato; comprendere i principali metodi. Introdurre i database
Allineamenti Multipli di Sequenze 1 Allineamento multiplo di sequenze: obiettivi di oggi Definire un allineamento multiplo di sequenze; com è generato; comprendere i principali metodi. Introdurre i database
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
Metodi euristici di allineamento
 Metodi euristici di allineamento Algoritmi euristici di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca rapida, anche se meno accurata, utilizzando la similarità
Metodi euristici di allineamento Algoritmi euristici di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca rapida, anche se meno accurata, utilizzando la similarità
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
Allineamenti multipli
 Allineamenti multipli Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte
Allineamenti multipli Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini)
") Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
Ricerche con BLAST (Laboratorio)
") Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Bioinformatica ed applicazioni di bioinformatica strutturale!
 Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Università degli studi di Pisa
 Università degli studi di Pisa Nicola Guido PATTERNHUNTER: Faster and More Sensitive. Homology Search Seminario: Bioinformatica a.a. 2008/2009 Contenuto della presentazione Introduzione Scenario PatternHunter
Università degli studi di Pisa Nicola Guido PATTERNHUNTER: Faster and More Sensitive. Homology Search Seminario: Bioinformatica a.a. 2008/2009 Contenuto della presentazione Introduzione Scenario PatternHunter
Programmazione dinamica
 rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
rogrammazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Biologia Molecolare Computazionale
 Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
Programmazione dinamica
 Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
InfoBioLab I ENTREZ. ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche
 InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
Pairwise Sequence Alignment BIOINFORMATICA. Corso di Laurea in Ingegneria Informatica e Biomedica. Università Magna Graecia Catanzaro
 Pairwise Sequence Alignment BIOINFORMATICA Corso di Laurea in Ingegneria Informatica e Biomedica Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento Esatto di Coppie
Pairwise Sequence Alignment BIOINFORMATICA Corso di Laurea in Ingegneria Informatica e Biomedica Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento Esatto di Coppie
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Definizioni
Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Definizioni
Corso di Bioinformatica. Docente: Dr. Antinisca DI MARCO
 Corso di Bioinformatica Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@univaq.it Analisi Filogenetica Gene Ancestrale duplicazione genica La filogenesi è lo studio delle relazioni evolutive tra
Corso di Bioinformatica Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@univaq.it Analisi Filogenetica Gene Ancestrale duplicazione genica La filogenesi è lo studio delle relazioni evolutive tra
Allineamento multiplo di sequenze
 Allineamento multiplo di sequenze Nicola Vitacolonna vitacolo@dimi.uniud.it http://www.dimi.uniud.it/~vitacolo Università degli Studi di Udine 16 aprile 2002 Two homologous sequences whisper... a full
Allineamento multiplo di sequenze Nicola Vitacolonna vitacolo@dimi.uniud.it http://www.dimi.uniud.it/~vitacolo Università degli Studi di Udine 16 aprile 2002 Two homologous sequences whisper... a full
Allineamento e similarità di sequenze
 Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
BLAST: Basic Local Alignment Search Tool
 BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni:
: Allineamento di sequenze (parte 1) Antonella Meloni:") Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione possibile [Jain
Riconoscimento e recupero dell informazione per bioinformatica Clustering Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione possibile [Jain
Alberi filogenetici. File: alberi_filogenetici.odp. Riccardo Percudani 02/03/04
 Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Definizioni preliminari
Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Definizioni preliminari
Informatica e biotecnologie
 Informatica e biotecnologie Ricerca di informazioni e analisi di sequenze CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Informatica e biotecnologie Strumenti per raccogliere
Informatica e biotecnologie Ricerca di informazioni e analisi di sequenze CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Informatica e biotecnologie Strumenti per raccogliere
Lezione 2: Allineamento di sequenze. BLAST e CLUSTALW
 Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Espansione: si parte da uno stato e applicando gli operatori (o la funzione successore) si generano nuovi stati.
 si generano nuovi stati.") CERCARE SOLUZIONI Generare sequenze di azioni. Espansione: si parte da uno stato e applicando gli operatori (o la funzione successore) si generano nuovi stati. Strategia di ricerca: ad ogni passo scegliere
CERCARE SOLUZIONI Generare sequenze di azioni. Espansione: si parte da uno stato e applicando gli operatori (o la funzione successore) si generano nuovi stati. Strategia di ricerca: ad ogni passo scegliere
Modello computazionale per la predizione di siti di legame per fattori di trascrizione
 Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
2.2 Alberi di supporto di costo ottimo
 . Alberi di supporto di costo ottimo Problemi relativi ad alberi hanno numerose applicazioni: progettazione di reti (comunicazione, teleriscaldamento,...) protocolli reti IP memorizzazione compatta di
. Alberi di supporto di costo ottimo Problemi relativi ad alberi hanno numerose applicazioni: progettazione di reti (comunicazione, teleriscaldamento,...) protocolli reti IP memorizzazione compatta di
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I UPGMA
 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I p4 UPGMA Clustering gerarchico in PERL Implementazione di un algoritmo di clustering Utilizzo
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I p4 UPGMA Clustering gerarchico in PERL Implementazione di un algoritmo di clustering Utilizzo
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
Intelligenza Artificiale
 Intelligenza Artificiale 17 Marzo 2005 Nome e Cognome: Matricola: ESERCIZIO N 1 Ricerca Cieca 5 punti 1.A) Elencare in modo ordinato i nodi (dell'albero sotto) che vengono scelti per l'espansione dalle
Intelligenza Artificiale 17 Marzo 2005 Nome e Cognome: Matricola: ESERCIZIO N 1 Ricerca Cieca 5 punti 1.A) Elencare in modo ordinato i nodi (dell'albero sotto) che vengono scelti per l'espansione dalle
METODI DELLA RICERCA OPERATIVA
 Università degli Studi di Cagliari FACOLTA' DI INGEGNERIA CORSO DI METODI DELLA RICERCA OPERATIVA Dott.ing. Massimo Di Francesco (mdifrance@unica.it) i i Dott.ing. Maria Ilaria Lunesu (ilaria.lunesu@unica.it)
Università degli Studi di Cagliari FACOLTA' DI INGEGNERIA CORSO DI METODI DELLA RICERCA OPERATIVA Dott.ing. Massimo Di Francesco (mdifrance@unica.it) i i Dott.ing. Maria Ilaria Lunesu (ilaria.lunesu@unica.it)
Algoritmi greedy. Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione
 Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
Allineamenti Multipli
 Allineamenti Multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Allineamenti Multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Corso di Programmazione
 II Accertamento del 27 Marzo 2001 / A Risolvi i seguenti esercizi, riporta le soluzioni in modo chiaro negli appositi riquadri e giustifica sinteticamente le risposte utilizzando i fogli protocollo. Cosa
II Accertamento del 27 Marzo 2001 / A Risolvi i seguenti esercizi, riporta le soluzioni in modo chiaro negli appositi riquadri e giustifica sinteticamente le risposte utilizzando i fogli protocollo. Cosa
ITI INFORMATICA: STATISTICA
 ITI INFORMATICA: STATISTICA INDICE:.INFERENZA STATISTICA.IL CAMPIONAMENTO CASUALE.LA PROGRAMMAZIONE LINEARE.IL CAMPIONAMENTO STATISTICO.DISTRIBUZIONI CAMPIONARIE.L ALGORITMO DEL SIMPLESSO INFERENZA STATISTICA
ITI INFORMATICA: STATISTICA INDICE:.INFERENZA STATISTICA.IL CAMPIONAMENTO CASUALE.LA PROGRAMMAZIONE LINEARE.IL CAMPIONAMENTO STATISTICO.DISTRIBUZIONI CAMPIONARIE.L ALGORITMO DEL SIMPLESSO INFERENZA STATISTICA
Metodi di identificazione
 Metodi di identificazione Metodo di identificazione LS per sistemi ARX Sia yt un processo ARX generico con parametri ignoti: S: yt= B z A z ut 1 1 A z et ota: scegliere ut 1 è la scelta più generica possibile,
Metodi di identificazione Metodo di identificazione LS per sistemi ARX Sia yt un processo ARX generico con parametri ignoti: S: yt= B z A z ut 1 1 A z et ota: scegliere ut 1 è la scelta più generica possibile,
Analisi della struttura primaria delle proteine
 Analisi della struttura primaria delle proteine Strumenti on-line La maggior parte degli strumenti per l analisi della struttura primaria si trovano on-line all indirizzo www.expasy.org Ottenere la sequenza
Analisi della struttura primaria delle proteine Strumenti on-line La maggior parte degli strumenti per l analisi della struttura primaria si trovano on-line all indirizzo www.expasy.org Ottenere la sequenza
Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
A GRAPH METHOD FOR KEYWORD-BASED SELECTION OF THE TOP-K DATABASES
 QUANG HIEU VU, BENG CHIN OOI, DIMITRIS PAPADIAS, ANTHONY K. H. TUNG A GRAPH METHOD FOR KEYWORD-BASED SELECTION OF THE TOP-K DATABASES Gruppo 9 Cacciari Alessandro, Franceschelli Alessio, Iuliani Matteo
QUANG HIEU VU, BENG CHIN OOI, DIMITRIS PAPADIAS, ANTHONY K. H. TUNG A GRAPH METHOD FOR KEYWORD-BASED SELECTION OF THE TOP-K DATABASES Gruppo 9 Cacciari Alessandro, Franceschelli Alessio, Iuliani Matteo
Allineamento multiplo
 Allineamento multiplo Allineamenti multipli Vs. allineamenti a coppie A 1: 2: 3: 4: 5: 6: B 1: 2: 3: 4: 5: 6: Significato biologico dell allineamento multiplo L allineamento multiplo riassume La storia
Allineamento multiplo Allineamenti multipli Vs. allineamenti a coppie A 1: 2: 3: 4: 5: 6: B 1: 2: 3: 4: 5: 6: Significato biologico dell allineamento multiplo L allineamento multiplo riassume La storia
Claudio Estatico Equazioni non-lineari
 Claudio Estatico (claudio.estatico@uninsubria.it) Equazioni non-lineari 1 Equazioni non-lineari 1) Equazioni non-lineari e metodi iterativi. 2) Metodo di bisezione, metodo regula-falsi. 3) Metodo di Newton.
Claudio Estatico (claudio.estatico@uninsubria.it) Equazioni non-lineari 1 Equazioni non-lineari 1) Equazioni non-lineari e metodi iterativi. 2) Metodo di bisezione, metodo regula-falsi. 3) Metodo di Newton.
Tool di allineamento multiplo a confronto
 di allineamento multiplo a confronto Bioinformatica a.a. 2007/08 Andrea Renieri Matteo Tanca Università di Pisa, Dipartimento di Informatica 11 Dicembre 2007 SCHEMA DELLA PRESENTAZIONE 1 INTRODUZIONE Definizione
di allineamento multiplo a confronto Bioinformatica a.a. 2007/08 Andrea Renieri Matteo Tanca Università di Pisa, Dipartimento di Informatica 11 Dicembre 2007 SCHEMA DELLA PRESENTAZIONE 1 INTRODUZIONE Definizione
Corso di Visione Artificiale. Texture. Samuel Rota Bulò
 Corso di Visione Artificiale Texture Samuel Rota Bulò Texture Le texture sono facili da riconoscere ma difficili da definire. Texture Il fatto di essere una texture dipende dal livello di scala a cui si
Corso di Visione Artificiale Texture Samuel Rota Bulò Texture Le texture sono facili da riconoscere ma difficili da definire. Texture Il fatto di essere una texture dipende dal livello di scala a cui si
Introduzione al Column Generation Caso di Studio: il Bin Packing Problem
 Introduzione al Column Generation Caso di Studio: il Bin Packing Problem November 15, 2014 1 / 26 Introduzione Il column generation è una metodologia che può essere usata per risolvere problemi di ottimizzazione
Introduzione al Column Generation Caso di Studio: il Bin Packing Problem November 15, 2014 1 / 26 Introduzione Il column generation è una metodologia che può essere usata per risolvere problemi di ottimizzazione
UNIVERSITÀ DEGLI STUDI ROMA TRE Corso di Studi in Ingegneria Informatica Ricerca Operativa 1 Seconda prova intermedia 17 giugno 2013
 A UNIVERSITÀ DEGLI STUDI ROMA TRE Corso di Studi in Ingegneria Informatica Ricerca Operativa Seconda prova intermedia 7 giugno 0 Nome: Cognome: Matricola: Orale /06/0 ore aula N Orale 0/07/0 ore aula N
A UNIVERSITÀ DEGLI STUDI ROMA TRE Corso di Studi in Ingegneria Informatica Ricerca Operativa Seconda prova intermedia 7 giugno 0 Nome: Cognome: Matricola: Orale /06/0 ore aula N Orale 0/07/0 ore aula N
Algoritmi greedy. Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione
 Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
Algoritmi greedy Gli algoritmi che risolvono problemi di ottimizzazione devono in genere operare una sequenza di scelte per arrivare alla soluzione Gli algoritmi greedy sono algoritmi basati sull idea
Problema. Equazioni non lineari. Metodo grafico. Teorema. Cercare la soluzione di
 Problema Cercare la soluzione di Equazioni non lineari dove Se è soluzione dell equazione, cioè allora si dice RADICE o ZERO della funzione Metodo grafico Graficamente si tratta di individuare l intersezione
Problema Cercare la soluzione di Equazioni non lineari dove Se è soluzione dell equazione, cioè allora si dice RADICE o ZERO della funzione Metodo grafico Graficamente si tratta di individuare l intersezione
Il Problema dell Albero Ricoprente Minimo (Shortest Spanning Tree - SST)
") Il Problema dell Albero Ricoprente Minimo (Shortest Spanning Tree - SST) È dato un grafo non orientato G=(V,E). Ad ogni arco e i E, i=1,,m, è associato un costo c i 0 7 14 4 10 9 11 8 12 6 13 5 17 3 2
Il Problema dell Albero Ricoprente Minimo (Shortest Spanning Tree - SST) È dato un grafo non orientato G=(V,E). Ad ogni arco e i E, i=1,,m, è associato un costo c i 0 7 14 4 10 9 11 8 12 6 13 5 17 3 2
Tecniche di riconoscimento statistico
 On AIR s.r.l. Tecniche di riconoscimento statistico Applicazioni alla lettura automatica di testi (OCR) Parte 9 Alberi di decisione Ennio Ottaviani On AIR srl ennio.ottaviani@onairweb.com http://www.onairweb.com/corsopr
On AIR s.r.l. Tecniche di riconoscimento statistico Applicazioni alla lettura automatica di testi (OCR) Parte 9 Alberi di decisione Ennio Ottaviani On AIR srl ennio.ottaviani@onairweb.com http://www.onairweb.com/corsopr
UNIVERSITÀ DEGLI STUDI DI PAVIA FACOLTÀ DI INGEGNERIA. Algoritmi
 UNIVERSITÀ DEGLI STUDI DI PAVIA FACOLTÀ DI INGEGNERIA Algoritmi Algoritmi classici Alcuni problemi si presentano con elevata frequenza e sono stati ampiamente studiati Ricerca di un elemento in un vettore
UNIVERSITÀ DEGLI STUDI DI PAVIA FACOLTÀ DI INGEGNERIA Algoritmi Algoritmi classici Alcuni problemi si presentano con elevata frequenza e sono stati ampiamente studiati Ricerca di un elemento in un vettore
Filogenesi molecolare
 Filogenesi molecolare Geni ortologhi e geni paraloghi Geni ortologhi: geni simili riscontrabili in organismi correlati tra loro. Il fenomeno della speciazione porta alla divergenza dei geni e quindi delle
Filogenesi molecolare Geni ortologhi e geni paraloghi Geni ortologhi: geni simili riscontrabili in organismi correlati tra loro. Il fenomeno della speciazione porta alla divergenza dei geni e quindi delle
Il progetto Genoma Umano è iniziato nel E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA.
 Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Progettazione di Algoritmi (4, 6, 9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo. Appello del 30 Gennaio 2019.
 Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo. Appello del 30 Gennaio 2019.") COGNOME: Nome: Progettazione di Algoritmi (4, 6, 9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo Appello del 30 Gennaio 2019 Attenzione: Inserire i propri dati nell apposito spazio soprastante
COGNOME: Nome: Progettazione di Algoritmi (4, 6, 9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo Appello del 30 Gennaio 2019 Attenzione: Inserire i propri dati nell apposito spazio soprastante
Esercizio da portare all orale
 Laboratorio di Informatica 2004/05 Corso di laurea in biotecnologie Esercizio da portare all orale Create subito una cartella che porti il vostro cognome. Fate attenzione a salvare tutti i vostri file
Laboratorio di Informatica 2004/05 Corso di laurea in biotecnologie Esercizio da portare all orale Create subito una cartella che porti il vostro cognome. Fate attenzione a salvare tutti i vostri file
q xi Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p i >0;
 Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Analisi Numerica. Debora Botturi ALTAIR. Debora Botturi. Laboratorio di Sistemi e Segnali
 Analisi Numerica ALTAIR http://metropolis.sci.univr.it Argomenti Rappresentazione di sistemi con variabili di stato; Tecniche di integrazione numerica Obiettivo: risolvere sistemi di equazioni differenziali
Analisi Numerica ALTAIR http://metropolis.sci.univr.it Argomenti Rappresentazione di sistemi con variabili di stato; Tecniche di integrazione numerica Obiettivo: risolvere sistemi di equazioni differenziali
Daniela Lera A.A
 Daniela Lera Università degli Studi di Cagliari Dipartimento di Matematica e Informatica A.A. 2016-2017 Problemi non lineari Definizione f : R R F : R n R m f (x) = 0 F(x) = 0 In generale si determina
Daniela Lera Università degli Studi di Cagliari Dipartimento di Matematica e Informatica A.A. 2016-2017 Problemi non lineari Definizione f : R R F : R n R m f (x) = 0 F(x) = 0 In generale si determina
Uso dell algoritmo di Quantizzazione Vettoriale per la determinazione del numero di nodi dello strato hidden in una rete neurale multilivello
 Tesina di Intelligenza Artificiale Uso dell algoritmo di Quantizzazione Vettoriale per la determinazione del numero di nodi dello strato hidden in una rete neurale multilivello Roberto Fortino S228682
Tesina di Intelligenza Artificiale Uso dell algoritmo di Quantizzazione Vettoriale per la determinazione del numero di nodi dello strato hidden in una rete neurale multilivello Roberto Fortino S228682
Complementi di Matematica e Calcolo Numerico A.A Laboratorio 9 - Equazioni non lineari
 Complementi di Matematica e Calcolo Numerico A.A. 2017-2018 Laboratorio 9 - Equazioni non lineari Data f : R R determinare α R tale che f(α) = 0 Le soluzioni di questo problema vengono dette radici o zeri
Complementi di Matematica e Calcolo Numerico A.A. 2017-2018 Laboratorio 9 - Equazioni non lineari Data f : R R determinare α R tale che f(α) = 0 Le soluzioni di questo problema vengono dette radici o zeri
Parte III: Algoritmo di Branch-and-Bound
 Parte III: Algoritmo di Branch-and-Bound Sia Divide et Impera z* = max {c T x : x S} (1) un problema di ottimizzazione combinatoria difficile da risolvere. Domanda: E possibile decomporre il problema (1)
Parte III: Algoritmo di Branch-and-Bound Sia Divide et Impera z* = max {c T x : x S} (1) un problema di ottimizzazione combinatoria difficile da risolvere. Domanda: E possibile decomporre il problema (1)
TECNOLOGIE INFORMATICHE MULTIMEDIALI
 TECNOLOGIE INFORMATICHE MULTIMEDIALI ««Gli errori sono necessari, utili come il pane e spesso anche belli: per esempio, la torre di Pisa.» (Gianni Rodari, Il libro degli errori) Prof. Giorgio Poletti giorgio.poletti@unife.it
TECNOLOGIE INFORMATICHE MULTIMEDIALI ««Gli errori sono necessari, utili come il pane e spesso anche belli: per esempio, la torre di Pisa.» (Gianni Rodari, Il libro degli errori) Prof. Giorgio Poletti giorgio.poletti@unife.it
Progettazione di Algoritmi (9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo. Appello del 27 Giugno 2018.
 Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo. Appello del 27 Giugno 2018.") COGNOME: Nome: Progettazione di Algoritmi (9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo Appello del 27 Giugno 2018 Attenzione: Inserire i propri dati nell apposito spazio soprastante
COGNOME: Nome: Progettazione di Algoritmi (9 CFU) Classe 3 (matricole congrue 2 modulo 3) Prof.ssa Anselmo Appello del 27 Giugno 2018 Attenzione: Inserire i propri dati nell apposito spazio soprastante
La codifica di sorgente
 Tecn_prog_sist_inform Gerboni Roberta è la rappresentazione efficiente dei dati generati da una sorgente discreta al fine poi di trasmetterli su di un opportuno canale privo di rumore. La codifica di canale
Tecn_prog_sist_inform Gerboni Roberta è la rappresentazione efficiente dei dati generati da una sorgente discreta al fine poi di trasmetterli su di un opportuno canale privo di rumore. La codifica di canale
Come si sceglie l algoritmo di allineamento? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione?
 Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Paolo Mogorovich
 Sistemi Informativi Territoriali Paolo Mogorovich www.di.unipi.it/~mogorov .4 Dati vettoriali - Tecniche di rappresentazione Un layer areale può essere rappresentato utilizzando diverse tecniche. Per esempio:
Sistemi Informativi Territoriali Paolo Mogorovich www.di.unipi.it/~mogorov .4 Dati vettoriali - Tecniche di rappresentazione Un layer areale può essere rappresentato utilizzando diverse tecniche. Per esempio:
Informatica e Bioinformatica A. A
 Purtroppo non esiste un modo univoco per indicare un gene. Ad esempio abbiamo visto che il gene tcap a seconda del record è riportato come titin-cap protein o telethonin. Questo crea confusione e non facilita
Purtroppo non esiste un modo univoco per indicare un gene. Ad esempio abbiamo visto che il gene tcap a seconda del record è riportato come titin-cap protein o telethonin. Questo crea confusione e non facilita
Corso di Bioinformatica
 Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
Programmazione Lineare: problema del trasporto Ing. Valerio Lacagnina
 Problemi di trasporto Consideriamo un problema di programmazione lineare con una struttura matematica particolare. Si può utilizzare, per risolverlo, il metodo del simplesso ma è possibile realizzare una
Problemi di trasporto Consideriamo un problema di programmazione lineare con una struttura matematica particolare. Si può utilizzare, per risolverlo, il metodo del simplesso ma è possibile realizzare una
Allineamenti di sequenze: concetti e algoritmi
 Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
OTTIMIZZAZIONE NON LINEARE
 OTTIMIZZAZIONE NON LINEARE In molti casi pratici non esistono algoritmi specifici per la soluzione del problema. Si utilizzano quindi algoritmi basati su approssimazioni locali della funzione o algoritmi
OTTIMIZZAZIONE NON LINEARE In molti casi pratici non esistono algoritmi specifici per la soluzione del problema. Si utilizzano quindi algoritmi basati su approssimazioni locali della funzione o algoritmi
1 TEORIA DELLE RETI 1. 1 Teoria delle reti. 1.1 Grafi
 1 TEORIA DELLE RETI 1 1 Teoria delle reti 1.1 Grafi Intuitivamente un grafo è un insieme finito di punti (nodi o vertici) ed un insieme di frecce (archi) che uniscono coppie di punti Il verso della freccia
1 TEORIA DELLE RETI 1 1 Teoria delle reti 1.1 Grafi Intuitivamente un grafo è un insieme finito di punti (nodi o vertici) ed un insieme di frecce (archi) che uniscono coppie di punti Il verso della freccia
Modelli di recupero. Modello di recupero booleano
 Modelli di recupero L obiettivo è recuperare i documenti che sono verosimilmente rilevanti all interrogazione. Vi sono vari modelli di recupero, che possono essere suddivisi in due grandi famiglie: exact
Modelli di recupero L obiettivo è recuperare i documenti che sono verosimilmente rilevanti all interrogazione. Vi sono vari modelli di recupero, che possono essere suddivisi in due grandi famiglie: exact
GESTIONE DELLA MEMORIA CENTRALE
 GESTIONE DELLA MEMORIA CENTRALE E MEMORIA VIRTUALE 7.1 Gestione della memoria Segmentazione Segmentazione con paginazione Memoria Virtuale Paginazione su richiesta Sostituzione delle pagine Trashing Esempi:
GESTIONE DELLA MEMORIA CENTRALE E MEMORIA VIRTUALE 7.1 Gestione della memoria Segmentazione Segmentazione con paginazione Memoria Virtuale Paginazione su richiesta Sostituzione delle pagine Trashing Esempi:
Problemi, istanze, soluzioni
 lgoritmi e Strutture di Dati II 2 Problemi, istanze, soluzioni Un problema specifica una relazione matematica tra dati di ingresso e dati di uscita. Una istanza di un problema è formata dai dati di un
lgoritmi e Strutture di Dati II 2 Problemi, istanze, soluzioni Un problema specifica una relazione matematica tra dati di ingresso e dati di uscita. Una istanza di un problema è formata dai dati di un
Intelligenza Artificiale. Clustering. Francesco Uliana. 14 gennaio 2011
 Intelligenza Artificiale Clustering Francesco Uliana 14 gennaio 2011 Definizione Il Clustering o analisi dei cluster (dal termine inglese cluster analysis) è un insieme di tecniche di analisi multivariata
Intelligenza Artificiale Clustering Francesco Uliana 14 gennaio 2011 Definizione Il Clustering o analisi dei cluster (dal termine inglese cluster analysis) è un insieme di tecniche di analisi multivariata
Algoritmi Greedy. Tecniche Algoritmiche: tecnica greedy (o golosa) Un esempio
 Un esempio") Algoritmi Greedy Tecniche Algoritmiche: tecnica greedy (o golosa) Idea: per trovare una soluzione globalmente ottima, scegli ripetutamente soluzioni ottime localmente Un esempio Input: lista di interi
Algoritmi Greedy Tecniche Algoritmiche: tecnica greedy (o golosa) Idea: per trovare una soluzione globalmente ottima, scegli ripetutamente soluzioni ottime localmente Un esempio Input: lista di interi
Complementi di Matematica e Calcolo Numerico A.A Laboratorio 6 Metodi iterativi per sistemi lineari
 Complementi di Matematica e Calcolo Numerico A.A. 2017-2018 Laboratorio 6 Metodi iterativi per sistemi lineari Dati una matrice A R N N non singolare e un vettore b R N, un metodo iterativo per la risoluzione
Complementi di Matematica e Calcolo Numerico A.A. 2017-2018 Laboratorio 6 Metodi iterativi per sistemi lineari Dati una matrice A R N N non singolare e un vettore b R N, un metodo iterativo per la risoluzione