Contratti e Accordi con parti terze in un sistema di Farmacovigilanza
|
|
|
- Marina Fiori
- 6 anni fa
- Visualizzazioni
Transcript

1 Contratti e Accordi con parti terze in un sistema di Farmacovigilanza Paolo Porcelli XXVI CONGRESSO NAZIONALE GIQAR Palermo, 26 maggio
2 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi nell industria farmaceutica NO Attualmente INTERESSI DIRETTI: 1.1 Impiego per una società: Ruolo esecutivo in una società farmaceutica 1.2 Impiego per una società: Ruolo guida nello sviluppo di un prodotto farmaceutico Da 0 a 3 anni precedenti * Paolo Porcelli, secondo il regolamento sul Conflitto di Interessi approvato dal CdA AIFA in data e pubblicato sulla Gazzetta Ufficiale del in accordo con la policy EMA /626261/2014 sulla gestione del conflitto di interessi dei membri dei Comitati Scientifici e degli esperti. N.B. Il compenso ricevuto per questo intervento è regolato dalla contrattazione collettiva. oltre 3 anni precedenti obbligatorio obbligatorio 1.3 Impiego per una società: altre attività facoltativo 2. Consulenza per una società facoltativo 3. Consulente strategico per una società facoltativo 4. Interessi finanziari facoltativo 5. Titolarità di un brevetto facoltativo INTERESSI INDIRETTI: 6. Sperimentatore principale facoltativo 7. Sperimentatore facoltativo 8. Sovvenzioni o altri fondi finanziari facoltativo 9. Interessi Familiari facoltativo

3 Argomenti Introduzione Riferimenti normativi Perché è necessario redigere contratti e accordi Differenti tipi di accordi: accordi di Co-marketing / Colicensing, distributori, produttori, outsourcing, Patient Support Programmes e Market Research Programmes, Investigator Initiated Studies, cambio di Titolarietà Deviazioni più comuni
")

")
 Dario Rovazzani")
4 Ufficio Ispezioni GVP Direttore Ufficio Ispezioni GVP (Elena Giovani) Assistente amministrativo Marisa Marchetti Ispettore Senior GVP Paolo Porcelli (farmacista) Ispettore Junior GVP Eraldo Donnarumma (farmacista) Candidato Ispettore Junior GVP Pina Guido (farmacista) Dario Rovazzani (medico) in distacco dalla CRI
5 Riferimenti normativi (1) Le disposizioni legislative di farmacovigilanza incluse nel Regolamento (CE) n. 726/2004, nella Direttiva 2001/83 / CE e nel Regolamento di esecuzione (UE) n. 520/2012 non comprendono definizioni giuridiche per i contratti e accordi (e statement of work). Non è prevista alcuna definizione di contratto e di accordo (e statement of work) nell Allegato I GVP - Definizioni (rev. 3). 5
6 Riferimenti normativi (2) Per quanto riguarda la collaborazione in materia di attività di farmacovigilanza, nel Regolamento di esecuzione (UE) n. 520/2012 (3) è riportato che il PSMF dovrebbe contenere informazioni chiave e documenti che coprono tutti gli aspetti delle attività di farmacovigilanza, comprese informazioni sulle attività che sono state subappaltate. Inoltre, nel caso di subappalto, l'articolo 6 del Regolamento di esecuzione (UE) n. 520/2012 della Commissione stati: 6
7 Perché è necessario avere contratti e accordi? Il titolare dell'autorizzazione all'immissione in commercio è tenuto ad avere contratti e accordi redatti in forma appropriata e coerente col sistema di farmacovigilanza adottato La QPPV è tenuta ad avere una sufficiente supervisione del funzionamento di questo sistema I contratti fanno parte del sistema di farmacovigilanza
8 L'importanza dei contratti Lo scopo della stesura di contratti / accordi di farmacovigilanza è per regolare i rapporti di lavoro tra le parti e in particolare per: facilitare la raccolta, la gestione e la protezione di dati di sicurezza fornire un quadro delle relazioni tra le parti dettagliare il trasferimento / delega di attività di farmacovigilanza descrivere la fornitura di servizi di farmacovigilanza da terze parti che comprendono i dettagli della supervisione delle attività da parte del titolare dell'autorizzazione all'immissione in commercio e gli standard di performance applicati GVP Module VI Section VI.C.2.2
 Co-marketing Co-licensing Patient Support Programmes")
9 Esempi dei più comuni tipi di accordi Distribution Manufacturing Outsourcing Change of ownership Market research programmes Investigator Initiated Studies (IIS) Co-marketing Co-licensing Patient Support Programmes (PSPs)
10 Accordi di Co-marketing / Co-licensing (1) Diversi in modo significativo a seconda del titolare di AIC o del medicinale: accordi di cooperazione allo sviluppo diversi titolari di licenza hanno una licenza per lo stesso medicinale in diversi territori diverse ditte commercializzano lo stesso medicinale con diversi nomi commerciali nello stesso territorio unico titolare di AIC, ma il titolare di AIC ha un accordo di vendita con un'altra azienda in uno o più territori 10
11 Accordi di Co-marketing / Co-licensing (2) GVP Module VI - Section IV.B.7: Where the MAH has set up contractual arrangements with a person or an organisation, explicit procedures and detailed agreements should exist between the MAH and the person/organisation to ensure that the MAH can comply with the reporting obligations. These procedures should in particular specify the processes for the exchange of safety information, including timelines and regulatory reporting responsibilities and should avoid duplicate reporting to competent authorities. 11
12 Accordi di Co-marketing / Co-licensing (3) Il titolare dell'autorizzazione all'immissione in commercio deve individuare correttamente tutti i dati che devono essere scambiati, le responsabilità, le modalità per gli scambi e le tempistiche. Si dovrà tener conto ad esempio di: AEs (Spontaneous, solicited, unsolicited) casi provenienti da MRP (Market Research Programmes) e PSP (Patient Support Program) dati di sicurezza relativi a situazioni particolari (ad esempio gravidanza, misuse, off-label use, ecc.) informazioni per l'adempimento di RMP misure (ad esempio follow-up mirati su eventi di particolare interesse) tempistiche e responsabilità definite per lo scambio delle informazioni di sicurezza specificare chiaramente se il titolare dell'autorizzazione all'immissione in commercio ha deciso di scambiare informazioni sui segnali 12
13 Accordi di Co-marketing / Co-licensing (4) Il titolare dell'autorizzazione all'immissione in commercio è responsabile del reference safety information? Produzione degli PSUR: l accordo specifica in che modo tutti i dati di sicurezza sono disponibili per essere inseriti negli PSURs? fa riferimento a un accordo sulle conclusioni degli PSUR? 13
14 Accordi di Co-marketing / Co-licensing (5) GVP Module VI - Section IV.B.4: When transfer of pharmacovigilance data occurs within an organisation or between organisations having concluded contractual agreements, the mechanism should be such that there is confidence that all notifications are received; in that, a confirmation and/or reconciliation process should be undertaken. C'è la garanzia che tutti i dati vengono raccolti e scambiati? In che modo ogni organizzazione avrà la supervisione del sistema? 14
15 Accordi di Co-marketing / Co-licensing problematiche/possibili deviazioni (1) Alcune attività di farmacovigilanza (le più significative) possono essere delegate ai partner di co-marketing / co-licensing e gestite quindi con i loro sistemi di farmacovigilanza. Tuttavia, nel PSMF si fa riferimento solo al contratto, invece che un' appropriata cross reference ai partner del PSMF. Mancanza di meccanismi appropriati per la supervisione delle attività di farmacovigilanza delegate dal titolare dell'autorizzazione all'immissione in commercio / meccanismi non chiaramente definiti anche quando i meccanismi sono descritti nell'accordo, essi non possono essere eseguiti (ad esempio la riconciliazione dei casi non viene effettuata con la frequenza definita). 15
16 Accordi di Co-marketing / Co-licensing problematiche/possibili deviazioni (2) Se un partner non detiene un'autorizzazione (MA) all'interno dell' EEA, il titolare dell'aic che invece la detiene nella EEA deve assicurare che gli accordi siano sufficientemente dettagliati per garantire che i requisiti di farmacovigilanza nell'eea siano soddisfatti e pensati con la dovuta e opportuna flessibilità. Problematiche già individuate dovrebbero essere incluse; scambio elettronico E2B; QC dei dati in entrata non tiene conto dei requisiti europei date di ricezione riportate in modo errato in riferimento al day zero; descrizione insufficiente del sistema di farmocovigilanza del partner nel PSMF del titolare di AIC EU per consentire a questo di essere conforme ai requisiti di farmacovigilanza della EEA. Un semplice riferimento nello SDEA non è appropriato. 16
17 Accordi di Co-marketing / Co-licensing problematiche/possibili deviazioni (3) Mancanza della previsione di modalità operative per la condivisione di audit findings relativi al sistema di farmacovigilanza del partner che influenza o può influenzare la performance delle attività di farmacovigilanza svolte per conto del titolare dell'autorizzazione all'immissione in commercio. 17
18 Distributori (1) A seconda del tipo di distributori, l'entità delle informazioni da inserire negli accordi possono variare; Distributori che sono titolari dell'autorizzazione all'immissione in commercio a livello nazionale a seconda delle disposizioni sulle responsabilità per lo scambio di informazione sul segnale, potrebbe essere necessario includere. Distributori che sono anche coinvolti nell implementazione di misure di minimizzazione dei rischi (ad esempio distribuzione di Materiali didattici) gli accordi devono prevedere disposizioni per garantire che il distributore venga informato degli aggiornamenti di questi materiali. 18
19 Distributori (2) Distributori che sono coinvolti solo nel trasporto dei medicinali senza alcun altra attività associata (ad es. grossisti) una richiesta di base sulle responsabilità per lo scambio di AEs e PQCs deve essere inclusa. 19
20 Produttori (1) Le informazioni sul produttore contenute nel contratto sono presenti sul PIL (o sull imballaggio esterno)? Percorso potenziale per la segnalazione di AE / reclami (complaints). C'è bisogno di descrivere il processo per il trasferimento di queste informazioni al titolare dell'autorizzazione all'immissione in commercio; definire la tipologia di informazioni da trasferire tempistiche attività di riconciliazione. 20
21 Produttori (2) Considerazione per i casi in cui il produttore è anche titolare di AIC per il principio attivo: c'è una accordo valido che regola lo scambio di informazioni su importanti problemi di sicurezza che potrebbero avere un impatto su entrambe le autorizzazioni? Deve essere previsto un accordo per garantire che il titolare dell'autorizzazione all'immissione in commercio sia informato di tutti gli eventuali concern di sicurezza / revisioni di qualità del prodotto (PQRs). Ciò potrebbe essere particolarmente rilevanti per i biologici. 21
22 Produttori - problematiche/possibili deviazioni Il produttore deve essere riportato sui PIL (imballaggio esterno?). Nell'accordo non è specificata la lingua da utilizzare per lo scambio di AE, casi da situazioni speciali e PQC Laddove la lingua da utilizzare relativamente allo scambio di informazioni sulla sicurezza è stata specificata, può mancare la conferma dei casi e / o la previsione di un processo di riconciliazione 22
23 Outsourcing Il livello delle informazioni incluse negli accordi può differire a seconda del tipo di attività affidata in outsourcing; è a livello globale o locale? è per tutte le attività del sistema di farmacovigilanza o solo per alcune attività (ad esempio preparazione degli PSUR, QPPV)? Le attività esternalizzate sono dettagliate in maniera adeguata? E stata prevista la condivisione dei risultati di ispezioni / audit? E stata prevista anche la condivisione di qualsiasi altra informazione (ad esempio SmPCs / PIL aggiornati)? 23
24 Outsourcing - problematiche/possibili deviazioni mancanza di meccanismi appropriati per la supervisione di tutte le attività di farmacovigilanza delegate dal titolare di AIC / meccanismi non chiaramente definiti meccanismi potrebbero includere indicatori di performance, controlli a campione / QC e audit mancata previsione della condivisione delle risultanze di audit / ispezioni relative al fornitore di servizi che influenzano o potrebbero influenzare le prestazioni delle attività di farmacovigilanza svolte da questo per conto del titolare dell'autorizzazione all'immissione in commercio mancata previsione relativa al modo in cui la parte terza deve essere tenuta aggiornata su informazioni necessarie allo svolgimento delle attività affidatele (ad esempio aggiornamenti a SmPCs (se è il caso), cambiamenti nelle SOP del titolare di AIC (quando questa utilizza le SOP del MAH) 24
25 Patient Support Programmes e Market Research Programmes E stato previsto qualcosa per lo scambio di specifiche informazioni sulla sicurezza con associate le relative tempistiche? Sono state previste le informazione che il titolare di AIC deve fornire alla parte terza (ad esempio materiali formativi, SmPC / PIL, FAQ)? Sono contemplate misure per assicurare che il personale che svolge le attività sia adeguatamente formato nei processi in generale e in particolare per la segnalazione di AE? Sono previste attività per la supervisione appropriata del titolare di AIC sui programmi? 25
26 Investigator Initiated Studies (IIS) (1) Le sospette reazioni avverse ai medicinali oggetto di studi clinici dovrebbero essere inoltrate solo dallo sponsor sulla base di quanto previsto per la sperimentazione clinica. Nel caso di IIS, lo sponsor deve essere l'investigator o l'istituzione associata. L 'accordo dovrebbe chiaramente definire chi è responsabile della segnalazione di SUSARs alle Autorità Competenti nazionali e ai comitati etici. La maggior parte degli ispettori/nca si aspettano almeno uno scambio bidirezionale tra il ricercatore e il titolare dell'autorizzazione all'immissione in commercio delle informazioni di sicurezza più importanti, che potrebbero influenzare la conduzione dello studio o il profilo beneficio/rischio del medicinale autorizzato. 26
27 Investigator Initiated Studies (IIS) (2) Se il titolare dell'autorizzazione all'immissione in commercio fornisce finanziamenti e / o il medicinale (gratuitamente), è necessario un accordo per prevedere almeno le aspettative/richieste minime. Sono consigliati lo scambio di SAE, interim safety analyses e final study reports. Queste informazioni devono essere fornite e considerate nei processi di analisi del segnale del titolare di AIC. 27
28 Cambio di Titolarietà La domanda di trasferimento della titolarità deve contenere il contratto di cessione tra la società attuale titolare dell autorizzazione all immissione in commercio e la società che ha acquisito il diritto al trasferimento della titolarità. Nel contratto deve essere esplicitamente dichiarato che il nuovo titolare di AIC è in possesso di: ICSRs PSURs RMP / problemi di sicurezza in corso / impegni post-marketing (commitments) segnali identificati e sotto valutazione variazioni di sicurezza pending L'accordo dovrebbe specificare come e quando i dati saranno trasferiti. 28
29 Cambio di Titolarietà - problematiche/possibili deviazioni Dati di farmacovigilanza non resi disponibili dal precedente proprietario / impegno del nuovo proprietario per recuperare i dati particolari problematiche per le quali il proprietario precedente è in liquidazione / fallimento variazioni di sicurezza pending o non presentate dal precedente o nuovo titolare di AIC tempestivamente ritardi nella presentazione di PSUR da parte del precedente titolare di AIC 29
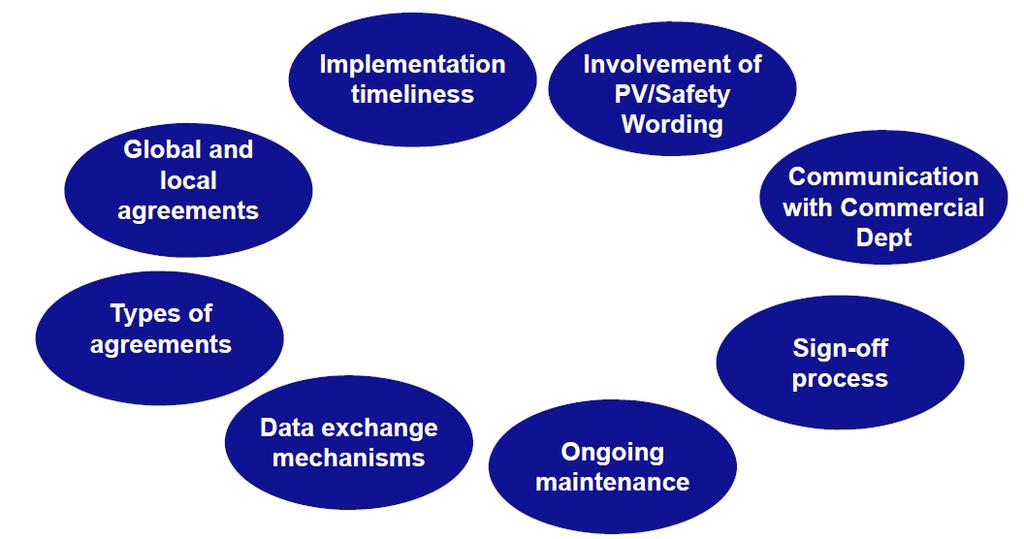
30 30
31 Deviazioni più comuni (1) Non è presente alcun accordo/contratto vigente Gli accordi/contratti sono scaduti Gli accordi/contratti non riflettono i requisiti legislativi vigenti Nessun processo di revisione è in atto 31
32 Deviazioni più comuni (2) Mancanza di oversight della QPPV e del dipartimento di farmacovigilanza sugli accordi contrattuali in vigore Mancato coinvolgimento della QPPV nella stesura e nell'approvazione di nuovi contratti Carenze negli accordi nessuna tempistica per lo scambio di dati/informazioni nessuna previsione per lo scambio di dati di sicurezza in situazioni speciali (ad esempio gravidanza, misuse, off-label use, etc ) esclusione di alcuni tipi di casi dallo scambio di dati 32

33 Phone: website:
La nuova legislazione sulla Farmacovigilanza. Il sistema di qualità di farmacovigilanza in AIFA. Pietro Erba. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza Il sistema di qualità di farmacovigilanza in AIFA Pietro Erba Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa
La nuova legislazione sulla Farmacovigilanza Il sistema di qualità di farmacovigilanza in AIFA Pietro Erba Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa
Ispezioni alle CRO. Angela Del Vecchio Direttore Ufficio Attività Ispettive GCP e di Farmacovigilanza
 Ispezioni alle CRO Angela Del Vecchio Direttore Ufficio Attività Ispettive GCP e di Farmacovigilanza XXV CONGRESSO NAZIONALE GIQAR Parma, 19 maggio 2016 Dichiarazione di trasparenza/interessi* Le opinioni
Ispezioni alle CRO Angela Del Vecchio Direttore Ufficio Attività Ispettive GCP e di Farmacovigilanza XXV CONGRESSO NAZIONALE GIQAR Parma, 19 maggio 2016 Dichiarazione di trasparenza/interessi* Le opinioni
La nuova legislazione sulla Farmacovigilanza. Il nuovo flusso delle segnalazioni. Carmela Santuccio. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza Il nuovo flusso delle segnalazioni Carmela Santuccio Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
La nuova legislazione sulla Farmacovigilanza Il nuovo flusso delle segnalazioni Carmela Santuccio Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
-La nuova legislazioni sulla farmacovigilanza -Analisi dei findings delle Ispezioni di Farmacovigilanza (Aprile Dicembre 2011)
") -La nuova legislazioni sulla farmacovigilanza -Analisi dei findings delle Ispezioni di Farmacovigilanza (Aprile 2010- Dicembre 2011) Angela Del Vecchio Dirigente Unità Ispezioni di Farmacovigilanza Roma,
-La nuova legislazioni sulla farmacovigilanza -Analisi dei findings delle Ispezioni di Farmacovigilanza (Aprile 2010- Dicembre 2011) Angela Del Vecchio Dirigente Unità Ispezioni di Farmacovigilanza Roma,
La nuova legislazione sulla Farmacovigilanza
 La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell Azienda Farmaceutica Alessandra Del Porto MSD Italia S.r.l., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma,
La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell Azienda Farmaceutica Alessandra Del Porto MSD Italia S.r.l., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma,
AIFA e studi di Fase I
 AIFA e studi di Fase I Sandra Petraglia 30 marzo 2016 Sperimentazione clinica di Fase I in Italia Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non
AIFA e studi di Fase I Sandra Petraglia 30 marzo 2016 Sperimentazione clinica di Fase I in Italia Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non
La nuova legislazione sulla Farmacovigilanza
 La nuova legislazione sulla Farmacovigilanza Il PSMF Il fascicolo di riferimento del sistema di farmacovigilanza Gian Nicola Castiglione - Chiesi Farmaceutici S.p.A., GdL Farmacovigilanza Farmindustria
La nuova legislazione sulla Farmacovigilanza Il PSMF Il fascicolo di riferimento del sistema di farmacovigilanza Gian Nicola Castiglione - Chiesi Farmaceutici S.p.A., GdL Farmacovigilanza Farmindustria
Farmacovigilanza e normativa: il punto di vista di un ispettore
 Farmacovigilanza e normativa: il punto di vista di un ispettore Nunzio Guido Mangano Verona, 23 settembre 2016 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono
Farmacovigilanza e normativa: il punto di vista di un ispettore Nunzio Guido Mangano Verona, 23 settembre 2016 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono
AUDIZIONE Camera dei Deputati. Malattie rare. Pierluigi Russo. Roma, 29 Aprile 2015
 AUDIZIONE Camera dei Deputati Malattie rare Pierluigi Russo Roma, 29 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
AUDIZIONE Camera dei Deputati Malattie rare Pierluigi Russo Roma, 29 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
02/11/2015. Nuovo Regolamento Europeo. Sandra Petraglia
 Nuovo Regolamento Europeo Sandra Petraglia 23 ottobre 2015 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi
Nuovo Regolamento Europeo Sandra Petraglia 23 ottobre 2015 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi
CASE STUDY: SISTEMA INTEGRATO ISO:9001-GVP
 CASE STUDY: SISTEMA INTEGRATO ISO:9001-GVP RAPPORTO PV SERVICE PROVIDER-TITOLARE AIC Dr.ssa Roberta Martorello QAPV SKILLPHARMA S.r.l. Via Umberto Saba 4-00144 Roma XXVI Congresso Nazionale GIQAR Palermo,
CASE STUDY: SISTEMA INTEGRATO ISO:9001-GVP RAPPORTO PV SERVICE PROVIDER-TITOLARE AIC Dr.ssa Roberta Martorello QAPV SKILLPHARMA S.r.l. Via Umberto Saba 4-00144 Roma XXVI Congresso Nazionale GIQAR Palermo,
USI CLINICI E SPERIMENTALI DI NUOVI RADIOFARMACI
 USI CLINICI E SPERIMENTALI DI NUOVI RADIOFARMACI Maria Nicotra 18 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
USI CLINICI E SPERIMENTALI DI NUOVI RADIOFARMACI Maria Nicotra 18 Aprile 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun
L Agenzia Italiana del Farmaco nel contesto europeo
 L Agenzia Italiana del Farmaco nel contesto europeo Luca Pani @Luca Pani - dg@aifa.gov.it Milano, 18 ottobre 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono
L Agenzia Italiana del Farmaco nel contesto europeo Luca Pani @Luca Pani - dg@aifa.gov.it Milano, 18 ottobre 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono
Le Nuove Buone Pratiche di Farmacovigilanza - SSFA
 Le Nuove Buone Pratiche di Farmacovigilanza - SSFA Roma, 23 gennaio 2013 Nuova legislazione di farmacovigilanza GVP Direttiva UE 84/2010 Regolamento UE 520/2012 Regolamento UE 1235/2010 Biggest legislative
Le Nuove Buone Pratiche di Farmacovigilanza - SSFA Roma, 23 gennaio 2013 Nuova legislazione di farmacovigilanza GVP Direttiva UE 84/2010 Regolamento UE 520/2012 Regolamento UE 1235/2010 Biggest legislative
Comitati etici: criticità e prospettive La posizione dell AIFA
 Comitati etici: criticità e prospettive La posizione dell AIFA Sandra Petraglia La Ricerca Clinica in Italia: evoluzione o involuzione? Bologna 7 novembre 2016 Dichiarazione di trasparenza/interessi* Le
Comitati etici: criticità e prospettive La posizione dell AIFA Sandra Petraglia La Ricerca Clinica in Italia: evoluzione o involuzione? Bologna 7 novembre 2016 Dichiarazione di trasparenza/interessi* Le
La nuova Legislazione sulla Farmacovigilanza Seminario Farmindustria in collaborazione con AIFA
 La nuova Legislazione sulla Farmacovigilanza Seminario Farmindustria in collaborazione con AIFA Roma, 13 settembre 2012 - Auditorium La nuova Confindustria, legislazione Viale sulla Tupini, Farmacovigilanza
La nuova Legislazione sulla Farmacovigilanza Seminario Farmindustria in collaborazione con AIFA Roma, 13 settembre 2012 - Auditorium La nuova Confindustria, legislazione Viale sulla Tupini, Farmacovigilanza
Algoritmo per la terapia dell osteoporosi. Mario Melazzini Roma, 11 novembre 2016
 Algoritmo per la terapia dell osteoporosi Mario Melazzini Roma, 11 novembre 2016 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
Algoritmo per la terapia dell osteoporosi Mario Melazzini Roma, 11 novembre 2016 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
Gli indicatori OsMed nella valutazione dell'appropriatezza d uso dei farmaci. Agnese Cangini 14 Luglio 2016
 Gli indicatori OsMed nella valutazione dell'appropriatezza d uso dei farmaci Agnese Cangini 14 Luglio 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
Gli indicatori OsMed nella valutazione dell'appropriatezza d uso dei farmaci Agnese Cangini 14 Luglio 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
Farmaci biologici nel trattamento della psoriasi: differenze nei criteri di valutazione tra USA e UE.
 Farmaci biologici nel trattamento della psoriasi: differenze nei criteri di valutazione tra USA e UE. Renato Bertini Malgarini, Giuseppe Pimpinella, Luca Pani 10 dicembre 2012- Istituto Superiore di Sanità
Farmaci biologici nel trattamento della psoriasi: differenze nei criteri di valutazione tra USA e UE. Renato Bertini Malgarini, Giuseppe Pimpinella, Luca Pani 10 dicembre 2012- Istituto Superiore di Sanità
La Sfida della Sostenibilità
 La Sfida della Sostenibilità Luca Pani Direttore Generale, AIFA DG@aifa.gov.it @Luca Pani Venezia, 31 Gennaio 2015 Dichiarazione di trasparenza/interessi* Interessi nell industria farmaceutica NO Attualmente
La Sfida della Sostenibilità Luca Pani Direttore Generale, AIFA DG@aifa.gov.it @Luca Pani Venezia, 31 Gennaio 2015 Dichiarazione di trasparenza/interessi* Interessi nell industria farmaceutica NO Attualmente
Appropriatezza prescrittiva, profili di utilizzo e aderenza ai farmaci d uso. Pierluigi Russo 21 Giugno 2016
 Appropriatezza prescrittiva, profili di utilizzo e aderenza ai farmaci d uso Pierluigi Russo 21 Giugno 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
Appropriatezza prescrittiva, profili di utilizzo e aderenza ai farmaci d uso Pierluigi Russo 21 Giugno 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali
La nuova legislazione sulla Farmacovigilanza
 La nuova legislazione sulla Farmacovigilanza Il Sistema di Gestione del Rischio Stefania De Santis Sigma-Tau IFR S.p.A., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma, 13 settembre 2012
La nuova legislazione sulla Farmacovigilanza Il Sistema di Gestione del Rischio Stefania De Santis Sigma-Tau IFR S.p.A., GdL Farmacovigilanza Farmindustria Auditorium Confindustria Roma, 13 settembre 2012
La nuova legislazione sulla Farmacovigilanza. I rapporti periodici di aggiornamento sulla sicurezza. Loriana Tartaglia. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza I rapporti periodici di aggiornamento sulla sicurezza Loriana Tartaglia Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse
La nuova legislazione sulla Farmacovigilanza I rapporti periodici di aggiornamento sulla sicurezza Loriana Tartaglia Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse
Good Clinical Practice (ICH- GCP): principi generali. Alessandra Foietta GMO Training & Compliance Manager Negrar 21 Ottobre 2016
: principi generali. Alessandra Foietta GMO Training & Compliance Manager Negrar 21 Ottobre 2016") Good Clinical Practice (ICH- GCP): principi generali Alessandra Foietta GMO Training & Compliance Manager Negrar 21 Ottobre 2016 Good Clinical Practice: cosa sono? Standard etico e di qualità internazionale
Good Clinical Practice (ICH- GCP): principi generali Alessandra Foietta GMO Training & Compliance Manager Negrar 21 Ottobre 2016 Good Clinical Practice: cosa sono? Standard etico e di qualità internazionale
funzioni di indirizzo vigilanza personalità giuridica di diritto pubblico e di autonomia organizzativa, patrimoniale, finanziaria e gestionale.
 AIFA. Art. 48,comma 13, del decreto-legge 30 settembre 2003,n.269,convertito nella legge 24 novembre 2003,n.326 Decreto 20 settembre 2004, n.245 Regolamento recante norme sull organizzazione ed il funzionamento
AIFA. Art. 48,comma 13, del decreto-legge 30 settembre 2003,n.269,convertito nella legge 24 novembre 2003,n.326 Decreto 20 settembre 2004, n.245 Regolamento recante norme sull organizzazione ed il funzionamento
Il Nuovo Regolamento Aziendale per. sperimentazioni. Collegio di Direzione Board Aziendale Ricerca e Innovazione 22 maggio 2013
 Il Nuovo Regolamento Aziendale per la conduzione di ricerche e sperimentazioni Collegio di Direzione Board Aziendale Ricerca e Innovazione 22 maggio 2013 DGR 1066/2009 Le funzioni della Ricerca Esplorativa/conoscitiva
Il Nuovo Regolamento Aziendale per la conduzione di ricerche e sperimentazioni Collegio di Direzione Board Aziendale Ricerca e Innovazione 22 maggio 2013 DGR 1066/2009 Le funzioni della Ricerca Esplorativa/conoscitiva
La nuova legislazione europea (84/2010)
") La nuova legislazione europea (84/2010) Fernanda Ferrazin Dirigente Ufficio di Farmacovigilanza Rimini, 30 Maggio 2012 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
La nuova legislazione europea (84/2010) Fernanda Ferrazin Dirigente Ufficio di Farmacovigilanza Rimini, 30 Maggio 2012 1 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
Angela Del Vecchio Direttore Ufficio Attività Ispettive GCP e di Farmacovigilanza. XXV CONGRESSO NAZIONALE GIQAR Parma, 19 maggio 2016
 Determina AIFA 2015 sui requisiti minimi necessari per le strutture sanitarie che eseguono sperimentazioni di fase I: scopi ed aspetti generali, parte clinica Angela Del Vecchio Direttore Ufficio Attività
Determina AIFA 2015 sui requisiti minimi necessari per le strutture sanitarie che eseguono sperimentazioni di fase I: scopi ed aspetti generali, parte clinica Angela Del Vecchio Direttore Ufficio Attività
Reazioni Avverse da Farmaco: Come e Perché segnalare
 CENTRO REGIONALE FARMACOVIGILANZA - Regione Sardegna UC di Farmacologia Clinica Azienda Ospedaliero Universitaria Cagliari Reazioni Avverse da Farmaco: Come e Perché segnalare Vaccini e Vaccinovigilanza
CENTRO REGIONALE FARMACOVIGILANZA - Regione Sardegna UC di Farmacologia Clinica Azienda Ospedaliero Universitaria Cagliari Reazioni Avverse da Farmaco: Come e Perché segnalare Vaccini e Vaccinovigilanza
DECRETI E DELIBERE DI ALTRE AUTORITÀ
 DECRETI E DELIBERE DI ALTRE AUTORITÀ DETERMINAZIONE 7 luglio 2010. AGENZIA ITALIANA DEL FARMACO Regime di rimborsabilità e prezzo di vendita del medicinale «Victoza». (Determinazione/C 397/2010). Regime
DECRETI E DELIBERE DI ALTRE AUTORITÀ DETERMINAZIONE 7 luglio 2010. AGENZIA ITALIANA DEL FARMACO Regime di rimborsabilità e prezzo di vendita del medicinale «Victoza». (Determinazione/C 397/2010). Regime
Le dimensioni del fenomeno e le iniziative dell AIFA
 Carenze dei farmaci: un fenomeno da contrastare Le dimensioni del fenomeno e le iniziative dell AIFA Domenico Di Giorgio Roma, 15 aprile 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse
Carenze dei farmaci: un fenomeno da contrastare Le dimensioni del fenomeno e le iniziative dell AIFA Domenico Di Giorgio Roma, 15 aprile 2016 Dichiarazione di trasparenza/interessi* Le opinioni espresse
Kit Documentale Qualità UNI EN ISO 9001:2015. Templates modificabili di Manuale, Procedure e Modulistica. Nuova versione 3.
 Premessa Il sistema di gestione per la qualità conforme alla norma internazionale UNI EN ISO 9001:2015 dovrebbe essere implementato nell ordine di seguito indicato, che riporta le clausole della norma
Premessa Il sistema di gestione per la qualità conforme alla norma internazionale UNI EN ISO 9001:2015 dovrebbe essere implementato nell ordine di seguito indicato, che riporta le clausole della norma
Bozza di Schema. Verso il Piano nazionale. sull'uso sostenibile dei prodotti fitosanitari. (documento strategico di sistema)
") Bozza di Schema Verso il Piano nazionale sull'uso sostenibile dei prodotti fitosanitari (documento strategico di sistema) 17 settembre 2009 1 Premessa Questo documento ha lo scopo di avviare la consultazione
Bozza di Schema Verso il Piano nazionale sull'uso sostenibile dei prodotti fitosanitari (documento strategico di sistema) 17 settembre 2009 1 Premessa Questo documento ha lo scopo di avviare la consultazione
DETERMINAZIONE 12 luglio Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010).
.") DETERMINAZIONE 12 luglio 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010). Regime di rimborsabilità e prezzo di vendita della specialità medicinale
DETERMINAZIONE 12 luglio 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onglyza». (Determinazione/C 412/2010). Regime di rimborsabilità e prezzo di vendita della specialità medicinale
Elementi di farmacovigilanza
 Elementi di farmacovigilanza Il Coordinamento della Ricerca 1 Corso ISG di Formazione per la Ricerca Clinica 28-Set-2015 Farmacovigilanza La sicurezza dei prodotti sperimentali (IMP) rappresenta un punto
Elementi di farmacovigilanza Il Coordinamento della Ricerca 1 Corso ISG di Formazione per la Ricerca Clinica 28-Set-2015 Farmacovigilanza La sicurezza dei prodotti sperimentali (IMP) rappresenta un punto
Corso di Laurea Magistrale in Chimica e Tecnologia Farmaceutiche E25
 Sezione di Tecnologia e Legislazione Farmaceutiche Maria Edvige Sangalli Corso di Laurea Magistrale in Chimica e Tecnologia Farmaceutiche E25 Fabbricazione Industriale dei Medicinali 4 CFU Farmacovigilanza
Sezione di Tecnologia e Legislazione Farmaceutiche Maria Edvige Sangalli Corso di Laurea Magistrale in Chimica e Tecnologia Farmaceutiche E25 Fabbricazione Industriale dei Medicinali 4 CFU Farmacovigilanza
Due nuove disposizioni in materia di farmacovigilanza
 Due nuove disposizioni in materia di farmacovigilanza Pubblicazione sulla Gazzetta Ufficiale Europea L 348 del 31 Dicembre 2010: DIRETTIVA UE 84/2010: modifica la Direttiva 2001/83/CE recante un codice
Due nuove disposizioni in materia di farmacovigilanza Pubblicazione sulla Gazzetta Ufficiale Europea L 348 del 31 Dicembre 2010: DIRETTIVA UE 84/2010: modifica la Direttiva 2001/83/CE recante un codice
Allegato I. Conclusioni scientifiche e motivazioni per la variazione dei termini dell autorizzazione delle autorizzazioni all immissione in commercio
 Allegato I Conclusioni scientifiche e motivazioni per la variazione dei termini dell autorizzazione delle autorizzazioni all immissione in commercio 1 Conclusioni scientifiche Tenendo conto della valutazione
Allegato I Conclusioni scientifiche e motivazioni per la variazione dei termini dell autorizzazione delle autorizzazioni all immissione in commercio 1 Conclusioni scientifiche Tenendo conto della valutazione
Questa policy riassume gli accordi in cui Novox Capital Ltd (di qui in avanti indicata quale la Compagnia )
") 1. Introduzione Questa policy riassume gli accordi in cui Novox Capital Ltd (di qui in avanti indicata quale la Compagnia ) si è impegnata secondo la Direttiva sugli Strumenti Finanziari ( MIFID ), la
1. Introduzione Questa policy riassume gli accordi in cui Novox Capital Ltd (di qui in avanti indicata quale la Compagnia ) si è impegnata secondo la Direttiva sugli Strumenti Finanziari ( MIFID ), la
Data: Novembre 2017 Presentazione: DIFETTI DI QUALITÀ IN MEDICINALI O MATERIE PRIME: A CHI, COME E QUANDO SEGNALARE
 Data: 13-15 Novembre 2017 Presentazione: DIFETTI DI QUALITÀ IN MEDICINALI O MATERIE PRIME: A CHI, COME E QUANDO SEGNALARE Ufficio Qualità dei Prodotti e Contrasto al Crimine Farmaceutico AIFA Dichiarazione
Data: 13-15 Novembre 2017 Presentazione: DIFETTI DI QUALITÀ IN MEDICINALI O MATERIE PRIME: A CHI, COME E QUANDO SEGNALARE Ufficio Qualità dei Prodotti e Contrasto al Crimine Farmaceutico AIFA Dichiarazione
Attività fisica ed esercizio fisico nella prevenzione e terapia del diabete. Alfonso Bellia
 Attività fisica ed esercizio fisico nella prevenzione e terapia del diabete Alfonso Bellia UNIVERSITA DI ROMA TOR VERGATA Dipartimento di Medicina dei Sistemi POLICLINICO TOR VERGATA Centro di riferimento
Attività fisica ed esercizio fisico nella prevenzione e terapia del diabete Alfonso Bellia UNIVERSITA DI ROMA TOR VERGATA Dipartimento di Medicina dei Sistemi POLICLINICO TOR VERGATA Centro di riferimento
Tutti i dati pubblicati nella Relazione ToV sono ad unico scopo di trasparenza e aderenza al Codice EFPIA sulla disclosure. Ogni altro uso dei dati
 Metodologia Daiichi Sankyo per la Pubblicazione dei Trasferimenti di Valore INDICE Privacy... 15 1. Consenso alla pubblicazione... 15 2. Consenso parziale... 15 CATEGORIE... 15 RICERCA E SVILUPPO... 15
Metodologia Daiichi Sankyo per la Pubblicazione dei Trasferimenti di Valore INDICE Privacy... 15 1. Consenso alla pubblicazione... 15 2. Consenso parziale... 15 CATEGORIE... 15 RICERCA E SVILUPPO... 15
Normativa Regolamento UE 1235/2010 Direttiva 2010/84/UE
 Implementazione delle nuove linee guida dell AIFA e principali modifiche introdotte dalla nuova normativa europea di Farmacovigilanza CEFPAS 24 marzo 2015 Claudia La Cavera Normativa Regolamento UE 1235/2010
Implementazione delle nuove linee guida dell AIFA e principali modifiche introdotte dalla nuova normativa europea di Farmacovigilanza CEFPAS 24 marzo 2015 Claudia La Cavera Normativa Regolamento UE 1235/2010
ORIENTAMENTI SU TEST, VERIFICHE O ESERCIZI CHE POSSONO PORTARE A MISURE DI SOSTEGNO ABE/GL/2014/ settembre 2014
 ABE/GL/2014/09 22 settembre 2014 Orientamenti sui tipi di test, verifiche o esercizi che possono portare a misure di sostegno ai sensi dell articolo 32, paragrafo 4, lettera d), punto iii), della direttiva
ABE/GL/2014/09 22 settembre 2014 Orientamenti sui tipi di test, verifiche o esercizi che possono portare a misure di sostegno ai sensi dell articolo 32, paragrafo 4, lettera d), punto iii), della direttiva
Dichiarazione di trasparenza/interessi*
 Il regolamento EU 536/2014: a che punto siamo Sandra Petraglia Il nuovo Regolamento Europeo: criticità e opportunità per la ricerca italiana Bologna 8 giugno 2016 Dichiarazione di trasparenza/interessi*
Il regolamento EU 536/2014: a che punto siamo Sandra Petraglia Il nuovo Regolamento Europeo: criticità e opportunità per la ricerca italiana Bologna 8 giugno 2016 Dichiarazione di trasparenza/interessi*
ALPHA SGR S.p.A. MANUALE DELLE PROCEDURE INTERNE PARTE GENERALE
 MANUALE DELLE PROCEDURE INTERNE PARTE GENERALE INDICE 1. Informazioni sulla Società ed attività autorizzate... 3 2. Autore del manuale delle procedure interne... 4 3. Finalità del manuale... 4 4. Contenuto
MANUALE DELLE PROCEDURE INTERNE PARTE GENERALE INDICE 1. Informazioni sulla Società ed attività autorizzate... 3 2. Autore del manuale delle procedure interne... 4 3. Finalità del manuale... 4 4. Contenuto
Sperimentazione Clinica: ruolo e responsabilità dell Infermiere
 Sperimentazione Clinica: ruolo e responsabilità dell Infermiere I diversi ruoli e responsabilità nella ricerca clinica Sergio Scaccabarozzi, Head of Clinical Operations-Roche S.p.A. Davide Ausili, RN,MSN,PhD.
Sperimentazione Clinica: ruolo e responsabilità dell Infermiere I diversi ruoli e responsabilità nella ricerca clinica Sergio Scaccabarozzi, Head of Clinical Operations-Roche S.p.A. Davide Ausili, RN,MSN,PhD.
SEGNALAZIONE DELLE SOSPETTE REAZIONI AVVERSE DA FARMACI. Un impegno etico per la. sicurezza del farmaco. Prevenire è meglio che curare
 Azienda U.S.L. 5 Spezzino U.O. Farmaceutica Territoriale Direttore Dr. Alessandro Sarteschi Prevenire è meglio che curare Le segnalazioni devono essere inviate a: Servizio Farmacovigilanza e Centro Documentazione
Azienda U.S.L. 5 Spezzino U.O. Farmaceutica Territoriale Direttore Dr. Alessandro Sarteschi Prevenire è meglio che curare Le segnalazioni devono essere inviate a: Servizio Farmacovigilanza e Centro Documentazione
10 anni di AIFA. Luca Pani Roma, 3 giugno 2013
 10 anni di AIFA Luca Pani Roma, 3 giugno 2013 Dichiarazione di trasparenza/interessi* Interessi nell industria farmaceutica NO Attualmente Precedenti 2 anni Da oltre 2 a 5 anni precedenti Oltre 5 anni
10 anni di AIFA Luca Pani Roma, 3 giugno 2013 Dichiarazione di trasparenza/interessi* Interessi nell industria farmaceutica NO Attualmente Precedenti 2 anni Da oltre 2 a 5 anni precedenti Oltre 5 anni
MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE
 UAE/AR/PFG ';?'65 /2013 CLASSIFICAZIONE Al SENSI DELL'ART. 12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli
UAE/AR/PFG ';?'65 /2013 CLASSIFICAZIONE Al SENSI DELL'ART. 12 COMMA 5 LEGGE 8 NOVEMBRE 2012 N. 189 DI MEDICINALI PER USO UMANO- APPROVATI CON PROCEDURA CENTRALIZZATA IL DIRETTORE GENERALE Visti gli articoli
DICHIARAZIONE PUBBLICA DI INTERESSI E IMPEGNO ALLA RISERVATEZZA DEI SOGGETTI CHE COLLABORANO ALLE ATTIVITA DEL CONSIGLIO SANITARIO REGIONALE
 DICHIARAZIONE PUBBLICA DI INTERESSI E IMPEGNO ALLA RISERVATEZZA DEI SOGGETTI CHE COLLABORANO ALLE ATTIVITA DEL CONSIGLIO SANITARIO REGIONALE Questo documento consiste di tre parti, i suoi Dettagli Personali,
DICHIARAZIONE PUBBLICA DI INTERESSI E IMPEGNO ALLA RISERVATEZZA DEI SOGGETTI CHE COLLABORANO ALLE ATTIVITA DEL CONSIGLIO SANITARIO REGIONALE Questo documento consiste di tre parti, i suoi Dettagli Personali,
Fabbricazione Industriale dei Medicinali
 Universita degli Studi di Milano Corso di Laurea in Chimica e Tecnologia Farmaceutiche Fabbricazione Industriale dei Medicinali slides Dott. Vittorino Ravelli Prof. Andrea Gazzaniga ORGANIZZAZIONE AZIENDALE
Universita degli Studi di Milano Corso di Laurea in Chimica e Tecnologia Farmaceutiche Fabbricazione Industriale dei Medicinali slides Dott. Vittorino Ravelli Prof. Andrea Gazzaniga ORGANIZZAZIONE AZIENDALE
Presentazione: Criticità nella produzione di medicinali privi di AIC.
 Presentazione: Criticità nella produzione di medicinali privi di AIC. Relatore: Dr. Giampiero Lorenti e Dr. Giuseppe Pimpinella Data: Rimini, 13 Giugno 2013 Dichiarazione di trasparenza/interessi* Le opinioni
Presentazione: Criticità nella produzione di medicinali privi di AIC. Relatore: Dr. Giampiero Lorenti e Dr. Giuseppe Pimpinella Data: Rimini, 13 Giugno 2013 Dichiarazione di trasparenza/interessi* Le opinioni
Regole e Qualità nell Industria Farmaceutica III incontro Nazionale dei Regulatory Affairs
 Regole e Qualità nell Industria Farmaceutica III incontro Nazionale dei Regulatory Affairs Legge Balduzzi: art. 11 Revisione del prontuario ed altre disposizioni dirette a favorire l impiego razionale
Regole e Qualità nell Industria Farmaceutica III incontro Nazionale dei Regulatory Affairs Legge Balduzzi: art. 11 Revisione del prontuario ed altre disposizioni dirette a favorire l impiego razionale
La gestione degli eventi avversi e dei SAE
 Oncology Business Unit - GMO La gestione degli eventi avversi e dei SAE Francesca Bamonte, GMO Study Manager Negrar 21 Ottobre 2016 Definizione di evento avverso (AE) Qualsiasi episodio sfavorevole di
Oncology Business Unit - GMO La gestione degli eventi avversi e dei SAE Francesca Bamonte, GMO Study Manager Negrar 21 Ottobre 2016 Definizione di evento avverso (AE) Qualsiasi episodio sfavorevole di
Risk Management in pratica
 Risk Management in pratica Raffaele Di Marzo I 50 anni di SSFA e la ricerca clinica in Italia Roma, 1 aprile 2014 1 Risk Management: Case study Risk Mangement-Tracking tool Uno strumento pratico aziendale
Risk Management in pratica Raffaele Di Marzo I 50 anni di SSFA e la ricerca clinica in Italia Roma, 1 aprile 2014 1 Risk Management: Case study Risk Mangement-Tracking tool Uno strumento pratico aziendale
SERVIZI SPECIALISTICI LEGALI E TECNICO- SISTEMISTICI PER ADEGUAMENTO ATTIVITÀ AL CODICE DELLA PRIVACY.
 SERVIZI SPECIALISTICI LEGALI E TECNICO- SISTEMISTICI PER ADEGUAMENTO ATTIVITÀ AL CODICE DELLA PRIVACY. Il presente documento ha lo scopo di illustrare in dettaglio i servizi legali ed informatici specialistici
SERVIZI SPECIALISTICI LEGALI E TECNICO- SISTEMISTICI PER ADEGUAMENTO ATTIVITÀ AL CODICE DELLA PRIVACY. Il presente documento ha lo scopo di illustrare in dettaglio i servizi legali ed informatici specialistici
Titolare A.I.C.: NOVARTIS EUROPHARM LTD
 DETERMINAZIONE 4 agosto 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onbrez Breezhaler» (indacaterolo) - autorizzata con procedura centralizzata europea dalla Commissione Europea.
DETERMINAZIONE 4 agosto 2010. Regime di rimborsabilità e prezzo di vendita del medicinale «Onbrez Breezhaler» (indacaterolo) - autorizzata con procedura centralizzata europea dalla Commissione Europea.
DETERMINAZIONE 25 marzo Regime di rimborsabilità e prezzo di vendita del medicinale per uso umano «Ranexa». (Determinazione n. 360/2010).
.") DETERMINAZIONE 25 marzo 2010. Regime di rimborsabilità e prezzo di vendita del medicinale per uso umano «Ranexa». (Determinazione n. 360/2010). Regime di rimborsabilità e prezzo di vendita della specialità
DETERMINAZIONE 25 marzo 2010. Regime di rimborsabilità e prezzo di vendita del medicinale per uso umano «Ranexa». (Determinazione n. 360/2010). Regime di rimborsabilità e prezzo di vendita della specialità
La nuova legislazione sulla Farmacovigilanza. Responsabilità e compiti dell AIFA. Laura Sottosanti. Roma, 13 settembre 2012
 La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell AIFA Laura Sottosanti Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
La nuova legislazione sulla Farmacovigilanza Responsabilità e compiti dell AIFA Laura Sottosanti Roma, 13 settembre 2012 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione
Regime di rimborsabilità e prezzo di vendita del medicinale PROLIA (denosumab). (Determinazione/C n. 2535/2011).
. (Determinazione/C n. 2535/2011).") DETERMINAZIONE 8 agosto 2011. Regime di rimborsabilità e prezzo di vendita del medicinale PROLIA (denosumab). (Determinazione/C n. 2535/2011). Regime di rimborsabilità e prezzo di vendita della specialità
DETERMINAZIONE 8 agosto 2011. Regime di rimborsabilità e prezzo di vendita del medicinale PROLIA (denosumab). (Determinazione/C n. 2535/2011). Regime di rimborsabilità e prezzo di vendita della specialità
Sistema di Farmacovigilanza: Interfacce tra PV e altre funzioni aziendali Introduzione al lavoro del GdLPV QA
 Sistema di Farmacovigilanza: Interfacce tra PV e altre funzioni aziendali Introduzione al lavoro del GdLPV QA XXIV GIQAR - Napoli, 27-29 Maggio 2015 Antonia Pascale R&D QA Unit Angelini in rappresentanza
Sistema di Farmacovigilanza: Interfacce tra PV e altre funzioni aziendali Introduzione al lavoro del GdLPV QA XXIV GIQAR - Napoli, 27-29 Maggio 2015 Antonia Pascale R&D QA Unit Angelini in rappresentanza
LA NUOVA LEGISLAZIONE SULLA FARMACOVIGILANZA
 Aderente a POSITION PAPER LA NUOVA LEGISLAZIONE SULLA FARMACOVIGILANZA LA POSIZIONE DI ASSOGENERICI GENNAIO 2013 Passeggiata di Ripetta, 22 00186 Roma T: +39 06 5960 5324 F: +39 06 5431 323 www.assogenerici.it
Aderente a POSITION PAPER LA NUOVA LEGISLAZIONE SULLA FARMACOVIGILANZA LA POSIZIONE DI ASSOGENERICI GENNAIO 2013 Passeggiata di Ripetta, 22 00186 Roma T: +39 06 5960 5324 F: +39 06 5431 323 www.assogenerici.it
-Applicazione della terminologia MeDRA alla codifica delle ADR -La FV negli studi osservazionali e nei registri dei farmaci sottoposti a monitoraggio
 Centro Regionale di Farmacovigilanza -Applicazione della terminologia MeDRA alla codifica delle ADR -La FV negli studi osservazionali e nei registri dei farmaci sottoposti a monitoraggio Seminario multi-tematico
Centro Regionale di Farmacovigilanza -Applicazione della terminologia MeDRA alla codifica delle ADR -La FV negli studi osservazionali e nei registri dei farmaci sottoposti a monitoraggio Seminario multi-tematico
BANDI RICERCA SCIENTIFICA
 F.A.Q. AGGIORNATE AL 08/04/2016 BANDI RICERCA SCIENTIFICA Bando con scadenza Ricerca biomedica condotta da giovani ricercatori 1. PRINCIPAL INVESTIGATOR DIPENDENTI A TEMPO INDETERMINATO E MODULISTICA -
F.A.Q. AGGIORNATE AL 08/04/2016 BANDI RICERCA SCIENTIFICA Bando con scadenza Ricerca biomedica condotta da giovani ricercatori 1. PRINCIPAL INVESTIGATOR DIPENDENTI A TEMPO INDETERMINATO E MODULISTICA -
PROCEDURA DI ACCESSO IN CONDIZIONI ORDINARIE
 MODELLO DI PERCORSO REGIONALE UNIFICATO PER L ACCESSO ALL UTILIZZO TERAPEUTICO DI MEDICINALI SOTTOPOSTI A SPERIMENTAZIONE CLINICA (EXPANDED-ACCESS/USO COMPASSIONEVOLE) Il protocollo da sottoporre all impresa
MODELLO DI PERCORSO REGIONALE UNIFICATO PER L ACCESSO ALL UTILIZZO TERAPEUTICO DI MEDICINALI SOTTOPOSTI A SPERIMENTAZIONE CLINICA (EXPANDED-ACCESS/USO COMPASSIONEVOLE) Il protocollo da sottoporre all impresa
Verso il Regolamento Europeo La proposta di AIFA
 Verso il Regolamento Europeo La proposta di AIFA Sandra Petraglia 13 novembre 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
Verso il Regolamento Europeo La proposta di AIFA Sandra Petraglia 13 novembre 2015 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in
Dipartimento Interaziendale Farmaceutico UO Farmacia Ospedaliera. La gestione delle segnalazione di reazione avversa (ADR) a farmaco
 a farmaco") Pag. 1/6 La gestione delle segnalazione di reazione avversa (ADR) a farmaco 1. Lista di distribuzione...1 2. Emissione...1 3. Scopo...1 4. Campo di applicazione...1 5. Riferimenti...2 6. Definizioni...2
Pag. 1/6 La gestione delle segnalazione di reazione avversa (ADR) a farmaco 1. Lista di distribuzione...1 2. Emissione...1 3. Scopo...1 4. Campo di applicazione...1 5. Riferimenti...2 6. Definizioni...2
Il sistema di gestione e controllo: funzioni e organismi responsabili
 Il sistema di gestione e controllo: funzioni e organismi responsabili Questo materiale didattico rientra nell ambito dei Percorsi e-learning di alta formazione specialistica del Progetto Esperi@ - Rafforzamento
Il sistema di gestione e controllo: funzioni e organismi responsabili Questo materiale didattico rientra nell ambito dei Percorsi e-learning di alta formazione specialistica del Progetto Esperi@ - Rafforzamento
La Normativa sui PASS. Antonio Baldassarre
 La Normativa sui PASS Antonio Baldassarre Contesto normativo Direttiva 2010/84/UE Regolamento (EC) 1235/2010 GVP Module VIII + Annex Reg. di Esecuzione della Commissione n 520/2012 Q&A su misure di transizione
La Normativa sui PASS Antonio Baldassarre Contesto normativo Direttiva 2010/84/UE Regolamento (EC) 1235/2010 GVP Module VIII + Annex Reg. di Esecuzione della Commissione n 520/2012 Q&A su misure di transizione
Informazione Formazione e Addestramento del Personale
 Società Italiana Banche degli Occhi Torino, 10 ottobre 2009 IV Corso di Formazione Informazione Formazione e Addestramento del Personale Fondazione Banca degli Occhi del Veneto Normativa di riferimento
Società Italiana Banche degli Occhi Torino, 10 ottobre 2009 IV Corso di Formazione Informazione Formazione e Addestramento del Personale Fondazione Banca degli Occhi del Veneto Normativa di riferimento
Impatto della nuova normativa europea di farmacovigilanza sugli studi osservazionali
 Impatto della nuova normativa europea di farmacovigilanza sugli studi osservazionali Reporting rules Matteo Laurita Longo 1 GdL Farmacovigilanza «Ernesto Montagna» Riferimenti Regolamento 1235/2010 in
Impatto della nuova normativa europea di farmacovigilanza sugli studi osservazionali Reporting rules Matteo Laurita Longo 1 GdL Farmacovigilanza «Ernesto Montagna» Riferimenti Regolamento 1235/2010 in
MANUALE SISTEMA DI GESTIONE INTEGRATO QUALITA E AMBIENTE
 Rev. N Pag. 1 a 25 MANUALE SISTEMA DI GESTIONE INTEGRATO QUALITA E AMBIENTE STATO APPROVAZIONE Rev. N Pag. 2 a 25 Realizzato da: Riesaminato da: Approvato da: Nome e cognome Firma Ruolo REVISIONI N pagine
Rev. N Pag. 1 a 25 MANUALE SISTEMA DI GESTIONE INTEGRATO QUALITA E AMBIENTE STATO APPROVAZIONE Rev. N Pag. 2 a 25 Realizzato da: Riesaminato da: Approvato da: Nome e cognome Firma Ruolo REVISIONI N pagine
Agenda. L accesso al farmaco e il suo impiego sicuro ed appropriato come strumento di difesa della salute. Agenzia Italiana del Farmaco
 Agenda L accesso al farmaco e il suo impiego sicuro ed appropriato come strumento di difesa della salute Eraldo Donnarumma obiettivo primario dell AIFA: sicurezza e appropriatezza del farmaco attività
Agenda L accesso al farmaco e il suo impiego sicuro ed appropriato come strumento di difesa della salute Eraldo Donnarumma obiettivo primario dell AIFA: sicurezza e appropriatezza del farmaco attività
DETERMINAZIONE 17 dicembre 2008
 DETERMINAZIONE 17 dicembre 2008 Regime di rimborsabilita' e prezzo di vendita del medicinale «Eucreas» (vildagliptin + metformina cloridrato), autorizzata con procedura centralizzata europea dalla Commissione
DETERMINAZIONE 17 dicembre 2008 Regime di rimborsabilita' e prezzo di vendita del medicinale «Eucreas» (vildagliptin + metformina cloridrato), autorizzata con procedura centralizzata europea dalla Commissione
ALLEGATO N. 2. mini-hta delle Tecnologie
 ALLEGATO N. 2 mini-hta delle Tecnologie DATI PRELIMINARI 1. Proponente Azienda Dipartimento- Struttura 2. Identificazione della tecnologia proposta Nome, tipo, campo di applicazione 3. La tecnologia proposta
ALLEGATO N. 2 mini-hta delle Tecnologie DATI PRELIMINARI 1. Proponente Azienda Dipartimento- Struttura 2. Identificazione della tecnologia proposta Nome, tipo, campo di applicazione 3. La tecnologia proposta
Studi FARMACOLOGICI PROFIT
 CEI FONDAZIONE PTV POLICLINICO TOR VERGATA SOP Procedure Operative Standard U.O.C. Farmacia clinica Studi FARMACOLOGICI PROFIT COMPILARE E ALLEGARE ALLA DOCUMENTAZIONE DELLO STUDIO L APPENDICE 2 ALLA DOMANDA
CEI FONDAZIONE PTV POLICLINICO TOR VERGATA SOP Procedure Operative Standard U.O.C. Farmacia clinica Studi FARMACOLOGICI PROFIT COMPILARE E ALLEGARE ALLA DOCUMENTAZIONE DELLO STUDIO L APPENDICE 2 ALLA DOMANDA
SPERIMENTAZIONE CLINICA DEI MEDICINALI DI TIPO COMMERCIALE
 SPERIMENTAZIONE CLINICA DEI MEDICINALI DI TIPO COMMERCIALE MODULISTICA A CURA DELLO SPERIMENTATORE RESPONSABILE E DEL DIRETTORE DELL UNITÀ OPERATIVA COINVOLTA Per ulteriori informazioni consultare la segreteria
SPERIMENTAZIONE CLINICA DEI MEDICINALI DI TIPO COMMERCIALE MODULISTICA A CURA DELLO SPERIMENTATORE RESPONSABILE E DEL DIRETTORE DELL UNITÀ OPERATIVA COINVOLTA Per ulteriori informazioni consultare la segreteria
Dichiarazione di trasparenza/interessi*
 Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi nell industria farmaceutica NO Attualmente Precedenti
Dichiarazione di trasparenza/interessi* Le opinioni espresse in questa presentazione sono personali e non impegnano in alcun modo l AIFA Interessi nell industria farmaceutica NO Attualmente Precedenti
Allegato I. Conclusioni scientifiche e motivazioni per la variazione dei termini delle autorizzazioni all immissione in commercio
 Allegato I Conclusioni scientifiche e motivazioni per la variazione dei termini delle autorizzazioni all immissione in commercio 1 Conclusioni scientifiche Tenendo conto della valutazione del Comitato
Allegato I Conclusioni scientifiche e motivazioni per la variazione dei termini delle autorizzazioni all immissione in commercio 1 Conclusioni scientifiche Tenendo conto della valutazione del Comitato
Ravenna 2010 La comunicazione ambientale. La norma ISO 14063: Linee Guida per la Comunicazione Ambientale
 Ing. Alessandra Archetti CESQA Centro Studi Qualità Ambiente CURA Consorzio Universitario di Ricerca Applicata c/o Dipartimento di Processi Chimici dell Ingegneria Università di Padova tel +39 049 8275539/5536
Ing. Alessandra Archetti CESQA Centro Studi Qualità Ambiente CURA Consorzio Universitario di Ricerca Applicata c/o Dipartimento di Processi Chimici dell Ingegneria Università di Padova tel +39 049 8275539/5536
Il nuovo Regolamento Europeo 536/2014 sulla sperimentazione clinica dei medicinali: quali opportunità?
 Evento formativo Data Manager; Bologna 13 Maggio 2016 Il nuovo Regolamento Europeo 536/2014 sulla sperimentazione clinica dei medicinali: quali opportunità? Carlo Tomino Coordinatore Ricerca e Sviluppo
Evento formativo Data Manager; Bologna 13 Maggio 2016 Il nuovo Regolamento Europeo 536/2014 sulla sperimentazione clinica dei medicinali: quali opportunità? Carlo Tomino Coordinatore Ricerca e Sviluppo
Obblighi di controllo dei Fornitori esterni. Rischio tecnologico
 Obblighi di dei Fornitori esterni Rischio tecnologico Area di 1. Gestione obsolescenza Garantire continue disposizioni di supporto Il Fornitore deve tempestivamente avvisare Barclays quando viene a conoscenza
Obblighi di dei Fornitori esterni Rischio tecnologico Area di 1. Gestione obsolescenza Garantire continue disposizioni di supporto Il Fornitore deve tempestivamente avvisare Barclays quando viene a conoscenza
DIREZIONE DIRITTI DI CITTADINANZA E COESIONE SOCIALE SETTORE CONSULENZA GIURIDICA, RICERCA E SUPPORTO ORGANISMI DI GOVERNO CLINICO BELVEDERE KATIA
 REGIONE TOSCANA DIREZIONE DIRITTI DI CITTADINANZA E COESIONE SOCIALE SETTORE CONSULENZA GIURIDICA, RICERCA E SUPPORTO ORGANISMI DI GOVERNO CLINICO Il Dirigente Responsabile: BELVEDERE KATIA Decreto non
REGIONE TOSCANA DIREZIONE DIRITTI DI CITTADINANZA E COESIONE SOCIALE SETTORE CONSULENZA GIURIDICA, RICERCA E SUPPORTO ORGANISMI DI GOVERNO CLINICO Il Dirigente Responsabile: BELVEDERE KATIA Decreto non
Overview sui punti cardine del sistema di FV in compliance con i nuovi requisiti normativi. Milano -15 novembre Docente:
 Basicof Pharma Education Center Il nuovo Sistema di Farmacovigilanza Overview sui punti cardine del sistema di FV in compliance con i nuovi requisiti normativi Milano -15 novembre 2012 Perché partecipare
Basicof Pharma Education Center Il nuovo Sistema di Farmacovigilanza Overview sui punti cardine del sistema di FV in compliance con i nuovi requisiti normativi Milano -15 novembre 2012 Perché partecipare
SPUNTI DI FARMACOVIGILANZA
 SPUNTI DI FARMACOVIGILANZA farmacoviilanza 10/14/2009 1 Farmacoviilanza Raccolta sistematica ed analisi di un insieme articolato di informazioni Coinvole a diversi livelli tutta la comunità: pazienti,
SPUNTI DI FARMACOVIGILANZA farmacoviilanza 10/14/2009 1 Farmacoviilanza Raccolta sistematica ed analisi di un insieme articolato di informazioni Coinvole a diversi livelli tutta la comunità: pazienti,
HIGH RESEARCH. Decreto Legislativo 24 giugno 2003 n 211
 srl Decreto Legislativo 24 giugno 2003 n 211 Dr, President- General Manager HIGH RESERACH srl AriSla, Milano 18 gennaio 2016 Dlgs 211 Art 1 Decreto si applica a sperimentazioni condotte in GCP Non si applica
srl Decreto Legislativo 24 giugno 2003 n 211 Dr, President- General Manager HIGH RESERACH srl AriSla, Milano 18 gennaio 2016 Dlgs 211 Art 1 Decreto si applica a sperimentazioni condotte in GCP Non si applica
Spirit Statement 2013 Definire gli item standard dei trial clinici
 Sperimentazioni Cliniche Nuove Sfide per i Comitati Etici Bologna, 7 novembre 2014 Spirit Statement 2013 Definire gli item standard dei trial clinici Corrado Iacono Comitato Etico Bologna-Imola Outline
Sperimentazioni Cliniche Nuove Sfide per i Comitati Etici Bologna, 7 novembre 2014 Spirit Statement 2013 Definire gli item standard dei trial clinici Corrado Iacono Comitato Etico Bologna-Imola Outline
sul supporto implicito per le operazioni di cartolarizzazione
 EBA/GL/2016/08 24/11/2016 Orientamenti sul supporto implicito per le operazioni di cartolarizzazione 1 1. Conformità e obblighi di comunicazione Status giuridico degli orientamenti 1. Il presente documento
EBA/GL/2016/08 24/11/2016 Orientamenti sul supporto implicito per le operazioni di cartolarizzazione 1 1. Conformità e obblighi di comunicazione Status giuridico degli orientamenti 1. Il presente documento
MEDICINALI ALLOPATICI
 ALLEGATO 1 CONVEGNI E CONGRESSI Parere favorevole allo svolgimento di manifestazioni che comportano, per l'impresa farmaceutica, un onere inferiore a 5.000,00 Parere favorevole allo svolgimento di manifestazioni
ALLEGATO 1 CONVEGNI E CONGRESSI Parere favorevole allo svolgimento di manifestazioni che comportano, per l'impresa farmaceutica, un onere inferiore a 5.000,00 Parere favorevole allo svolgimento di manifestazioni
Non tutto è scritto sulla pietra!
 VI CORSO DI AGGIORNAMENTO SUI FARMACI: gli scenari che cambiano Cosa sta cambiando nella Farmacovigilanza? - esperienze in casa madre Nadia Canova (Alfa Wassermann SpA) GdL Farmacovigilanza SSFA Non tutto
VI CORSO DI AGGIORNAMENTO SUI FARMACI: gli scenari che cambiano Cosa sta cambiando nella Farmacovigilanza? - esperienze in casa madre Nadia Canova (Alfa Wassermann SpA) GdL Farmacovigilanza SSFA Non tutto
GMP nella produzione dei cosmetici
 GMP nella produzione dei cosmetici Giulia Trovato DIPARTIMENTO DI PREVENZIONE S.C. IGIENE E SANITA PUBBLICA ASL TO 5 Regolamento (CE) n.1223/2009 Considerazione 16 Per garantirne la sicurezza, i prodotti
GMP nella produzione dei cosmetici Giulia Trovato DIPARTIMENTO DI PREVENZIONE S.C. IGIENE E SANITA PUBBLICA ASL TO 5 Regolamento (CE) n.1223/2009 Considerazione 16 Per garantirne la sicurezza, i prodotti
Di Renzo Regulatory Affairs
 Di Renzo Regulatory Affairs Nel 1985, Di Renzo Regulatory Affairs ha iniziato la sua attività di consulenza regolatoria per i medicinali ad uso umano e veterinario, gli integratori alimentari, i cosmetici,
Di Renzo Regulatory Affairs Nel 1985, Di Renzo Regulatory Affairs ha iniziato la sua attività di consulenza regolatoria per i medicinali ad uso umano e veterinario, gli integratori alimentari, i cosmetici,
AIFA Agenzia Italiana del Farmaco Benfluorex
 AIFA - Benfluorex 18/12/2009 (Livello 2) AIFA Agenzia Italiana del Farmaco Benfluorex Comunicato EMEA Domande e Risposte (FAQ) file:///c /documenti/ben950.htm [18/12/2009 15.50.04] 18 December 2009 EMA/CHM/815033/2009
AIFA - Benfluorex 18/12/2009 (Livello 2) AIFA Agenzia Italiana del Farmaco Benfluorex Comunicato EMEA Domande e Risposte (FAQ) file:///c /documenti/ben950.htm [18/12/2009 15.50.04] 18 December 2009 EMA/CHM/815033/2009
Ufficio Studi Sonia Ciccolella e Mattia Suardi Proposta di Regolamento delegato, Direttiva IDD consultazione della Commissione europea
 Proposta di Regolamento delegato, Direttiva IDD consultazione della Commissione europea Premessa. La Commissione europea ha aperto una consultazione (scadenza: 17 agosto 2017) dedicata alla bozza di Regolamento
Proposta di Regolamento delegato, Direttiva IDD consultazione della Commissione europea Premessa. La Commissione europea ha aperto una consultazione (scadenza: 17 agosto 2017) dedicata alla bozza di Regolamento
Normativa recente in Farmacovigilanza
 Normativa recente in Farmacovigilanza Dipartimento Farmaceutico Asl RE marzo 2013 Elisa Iori 1 Attuale legislazione in vigore in tema di farmacovigilanza DECRETO LEGISLATIVO 24 aprile 2006, n. 219. Attuazione
Normativa recente in Farmacovigilanza Dipartimento Farmaceutico Asl RE marzo 2013 Elisa Iori 1 Attuale legislazione in vigore in tema di farmacovigilanza DECRETO LEGISLATIVO 24 aprile 2006, n. 219. Attuazione
(D.M.. 17 dicembre 2004)
") DICHIARAZIONE DI SPERIMENTAZIONI CLINICHE NON-PROFIT (D.M.. 17 dicembre 2004) Da compilarsi da parte di: Promotore (*): Sperimentatore (*): Studio : EUDRACT (nel caso di sperimentazioni col farmaco) Titolo
DICHIARAZIONE DI SPERIMENTAZIONI CLINICHE NON-PROFIT (D.M.. 17 dicembre 2004) Da compilarsi da parte di: Promotore (*): Sperimentatore (*): Studio : EUDRACT (nel caso di sperimentazioni col farmaco) Titolo
Legge federale sugli agenti terapeutici: nuovo quadro legislativo
 Serata AFTI Legge federale sugli agenti terapeutici: nuovo quadro legislativo F. Dotto 07 febbraio 2002 Ispettorato chimico-farmaceutico della Repubblica e Cantone Ticino 1 Riferimenti legislativi principali
Serata AFTI Legge federale sugli agenti terapeutici: nuovo quadro legislativo F. Dotto 07 febbraio 2002 Ispettorato chimico-farmaceutico della Repubblica e Cantone Ticino 1 Riferimenti legislativi principali
REGOLAMENTO (UE) N. 1078/2012 DELLA COMMISSIONE
 N. 1078/2012 DELLA COMMISSIONE") L 320/8 Gazzetta ufficiale dell Unione europea 17.11.2012 REGOLAMENTO (UE) N. 1078/2012 DELLA COMMISSIONE del 16 novembre 2012 relativo a un metodo di sicurezza comune per il monitoraggio che devono applicare
L 320/8 Gazzetta ufficiale dell Unione europea 17.11.2012 REGOLAMENTO (UE) N. 1078/2012 DELLA COMMISSIONE del 16 novembre 2012 relativo a un metodo di sicurezza comune per il monitoraggio che devono applicare
GESTIONE E CONTROLLO DEI DOCUMENTI E DELLE REGISTRAZIONI
 PER LA QUALITA 04 03.01.2009 1 6 GESTIONE E CONTROLLO DEI DOCUMENTI E DELLE REGISTRAZIONI 1. Scopo 2. Generalità 3. Identificazione 4. Emissione 5. Distribuzione 6. Archiviazione 7. Modifica 8. Controllo
PER LA QUALITA 04 03.01.2009 1 6 GESTIONE E CONTROLLO DEI DOCUMENTI E DELLE REGISTRAZIONI 1. Scopo 2. Generalità 3. Identificazione 4. Emissione 5. Distribuzione 6. Archiviazione 7. Modifica 8. Controllo