Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
|
|
|
- Franca Marrone
- 6 anni fa
- Visualizzazioni
Transcript
1 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
2 Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni Proteina vs. traduzione in aa di sequenze nucleotidiche (tutti i moduli) Query: DNA Protein Database: DNA Protein
3 Quando confrontiamo sequenze proteiche cerchiamo la migliore corrispondenza per 20 diversi amminoacidi Quando confrontiamo sequenze nucleotidiche cerchiamo la migliore corrispondenza per sole 4 basi nucleotidiche
4 Quando confrontiamo sequenze proteiche cerchiamo la migliore corrispondenza per 20 diversi amminoacidi Quando confrontiamo sequenze nucleotidiche cerchiamo la migliore corrispondenza per sole 4 basi nucleotidiche La probabilita di trovare una buona corrispondenza (allineamento con punteggio alto) per caso è più alta per le sequenze nucleotidiche che per quelle proteiche Inoltre, quando confrontiamo sequenze proteiche possiamo tener conto della similarita tra i diversi amminoacidi
5 Quando confrontiamo sequenze proteiche cerchiamo la migliore corrispondenza per 20 diversi amminoacidi Quando confrontiamo sequenze nucleotidiche cerchiamo la migliore corrispondenza per sole 4 basi nucleotidiche La probabilita di trovare una buona corrispondenza (allineamento con punteggio alto) per caso è più alta per le sequenze nucleotidiche che per quelle proteiche Inoltre, quando confrontiamo sequenze proteiche possiamo tener conto della similarita tra i diversi amminoacidi Quando è possibile, è preferibile confrontare sequenze proteiche!
6 Quante sequenze di proteine nelle banche dati? 800, , , , , , , , ,000 12, Protein sequences Globular 3D structures Membrane 3D structures
7 Come possiamo pescare dai databases di sequenze potenziali omologhe?
8 Algoritmi esatti (Smith-Waterman) Lezione precedente Esatto, garantisce di trovare il/i miglior allineamento/i per una coppia di sequenze. Per 2 sequenze: A di lunghezza n and B di lunghezza m, Smith-Waterman inpiega n*m passi computazionali. Cerchiamo omologhe della sequenza query A (n=200 aa) Cerchiamo nel DB (10 6 sequenze di m=200 aa) Numero di passi computazionali = 10 6 x 200 x 200 = ~ passi al sec = 10 7 secs = 120 giorni = 4 mesi! Necessità di algoritmi approssimati
9 Algoritmi esatti (Smith-Waterman) Lezione precedente Esatto, garantisce di trovare il/i miglior allineamento/i per una coppia di sequenze. Per 2 sequenze: A di lunghezza n and B di lunghezza m, Smith-Waterman inpiega n*m passi computazionali. Nel cercare Come in un scartiamo database gli di allineamenti sequenze- irrilevanti? - Nel caso la sequenza query A sia lunga n=200 nucleotidi - Cerchiamo sequenze omologhe nel database EST che contiene, e.g., 23*10 6 sequenze, B i, ciascuna Gli algoritmi di lunghezza euristici m=500. (BLAST, FASTA) servono a scartare - Numero di passi computazionali: la gran parte degli allineamenti irrilevanti. 23*10 6 * 500 * 200 ~ passi totali!
10 Programmi quali FASTA e BLAST, partendo da una sequenza query:
11 Programmi quali FASTA e BLAST, partendo da una sequenza query: prima pescano dalle banche dati un sottoinsieme di sequenze che sono potenziali omologhe

12 Programmi quali FASTA e BLAST, partendo da una sequenza query: prima pescano dalle banche dati un sottoinsieme di sequenze che sono potenziali omologhe poi allineano al meglio ciascuna sequenza di questo sottoinsieme alla sequenza query
13 FASTA: esempio query ACDDEFGSATRMASTRK Banca dati (DB) Seq 1: LKDCDDAFSGSTLTLMRASRK Seq 2: ACKRAEFSGSVTRMLSTRK Seq 3: ACDDEFGLLLTRYTMASTRK Step 1 = Divisione della sequenza in parole di 2 caratteri. Parole possibili: AC, CD, DD, DE, EF, FG, GS, SA, AT, TR, RM, MA, AS, ST, RK
14 Step 2 = Tabella delle frequenze delle parole Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Parola Query Seq 1 Seq 2 Seq 3 Off1 Off2 Off3 AC CD DD DE EF FG GS SA AT TR 10-12, , 7 1 RM MA AS ST RK
15 Step 3 = Calcolo dello score di similarita Init1 (basato sulla tabella dello step 2) Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Query A C D D E F G S A T R M A S T R K Pos X X X X X X X X Seq2 A C K R A E F S G S V T R M L S T R K T Pos Off Init1(seq2) basato su questo allineamento approssimato usando una matrice tipo PAM250
16 Step 3 = Calcolo dello score di similarita Init1 (basato sulla tabella dello step 2) Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Query Pos A C D D E F G S A T R M A S T R K X X X X X X Seq2 A C D D E F G L L L T R Y T M A S T R K Pos Off Init1(seq3) basato su questo allineamento approssimato usando una matrice tipo PAM250
17 Step 3 = Calcolo dello score di similarita Init1 (basato sulla tabella dello step 2) Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Query Pos A C D D E F G S A T R M A S T R K X X X X X X Seq2 A C D D E F G L L L T R Y T M A S T R K Pos Off Init1(seq3) basato su allineamento sia con off 0 che off 1 (nel caso di proteine lunghe si prendono i migliori 10 offs)
18 Sulla base degli score di similarita Init1 selezionare un sottoinsieme (grande) di sequenze per l analisi successiva Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Per calcolare Init1 si scelgono solo le 10 regioni con il maggiore allineamento
19 Sulla base degli score di similarita Init1 selezionare un sottoinsieme (grande) di sequenze per l analisi successiva Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK In questo modo abbiamo selezionato omologhe senza permettere inserzioni o delezioni. Introduzione dello score di similarità InitN.
20 Tabella delle frequenze delle parole Query: ACDDEFGSATRMASTRK Parola Query Seq 1 Seq 2 Seq 3 Off1 Off2 Off3 AC CD DD DE EF FG GS SA AT TR 10-12, , 7 1 RM MA AS ST RK
21 Step 4 = Calcolo dello score di similarita InitN (basato sull allineamento dello step 3 e sulla tabella dello step 2) Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Query A C - D D E F - G S A T R M A S T R K Pos X X X X X X X X X X X X Seq2 A C K R A E F S G S V T R M L S T R K T Pos Off InitN(seq2) = Init1(seq2) + score(nuovi matchs) K(gap)
22 Sulla base degli score di similarita InitN selezionare un sottoinsieme ridotto di sequenze per l analisi successiva Query: ACDDEFGSATRMASTRK DB: Seq 1 LKDCDDAFSGSTLTLMRASRK Seq 2 ACKRAEFSGSVTRMLSTRK Seq 3 ACDDEFGLLLTRYTMASTRK Step 5 = Allineamento vero e proprio delle sequenze con il miglior InitN alla query e calcolo del coefficiente finale opt (punteggio per il nuovo allineamento, completo) NOTA. La scelta del sottoinsieme di sequenze nel DB con cui si effettua l allineamento ottimale è basata sui punteggi approssimati Init1 e InitN
23 Algoritmo di FASTA 1. Suddivide le sequenze in parole (2 aa) 2. Trova le parole nelle sequenze del DB e calcola l offset 3. Calcola la similarita delle dieci regioni con maggiori parole identiche per ciascuna sequenza del DB (init1). Prima esclusione di sequenze 4. Calcola la similarita delle dieci regioni con maggiori parole identiche includendo gaps (initn) Seconda esclusione di sequenze 5. Allinea accuratamente le sequenze rimaste (opt) init1 initn opt
24 Quanto e buono un allineamento?
25 Quanto e buono un allineamento? = Quanto e migliore di uno casuale? Sequenze che danno un allineamento casuale: Sequenze non omologhe Sequenze rimescolate ( shuffled ) Sequenze generate casualmente Sequenze a bassa complessità
26 bassa complessità MMSFVQKGSW LLLALLHPTI ILAQQEAVEG GCSHLGQSYA DRDVWKPEPC QICVCDSGSV LCDDIICDDQ ELDCPNPEIP FGECCAVCPQ PPTAPTRPPN GQGPQGPKGD PGPPGIPGRN GDPGIPGQPG SPGSPGPPGI CESCPTGPQN YSPQYDSYDV KSGVAVGGLA GYPGPAGPPG PPGPPGTSGH PGSPGSPGYQ GPPGEPGQAG PSGPPGPPGA IGPSGPAGKD GESGRPGRPG ERGLPGPPGI KGPAGIPGFP GMKGHRGFDG RNGEKGETGA PGLKGENGLP GENGAPGPMG PRGAPGERGR PGLPGAAGAR GNDGARGSDG QPGPPGPPGT AGFPGSPGAK GEVGPAGSPG SNGAPGQRGE PGPQGHAGAQ GPPGPPGING SPGGKGEMGP AGIPGAPGLM GARGPPGPAG ANGAPGLRGG AGEPGKNGAK GEPGPRGERG EAGIPGVPGA KGEDGKDGSP GEPGANGLPG AAGERGAPGF RGPAGPNGIP GEKGPAGERG APGPAGPRGA AGEPGRDGVP GGPGMRGMPG SPGGPGSDGK PGPPGSQGES GRPGPPGPSG PRGQPGVMGF PGPKGNDGAP GKNGERGGPG GPGPQGPPGK NGETGPQGPP GPTGPGGDKG DTGPPGPQGL QGLPGTGGPP GENGKPGEPG PKGDAGAPGA PGGKGDAGAP GERGPPGLAG APGLRGGAGP PGPEGGKGAA GPPGPPGAAG TPGLQGMPGE RGGLGSPGPK GDKGEPGGPG ADGVPGKDGP RGPTGPIGPP GPAGQPGDKG EGGAPGLPGI AGPRGSPGER GETGPPGPAG FPGAPGQNGE PGGKGERGAP GEKGEGGPPG VAGPPGGSGP AGPPGPQGVK GERGSPGGPG AAGFPGARGL PGPPGSNGNP GPPGPSGSPG KDGPPGPAGN 0.3 TGAPGSPGVS GPKGDAGQPG EKGSPGAQGP PGAPGPLGIA GITGARGLAG PPGMPGPRGS PGPQGVKGES GKPGANGLSG ERGPPGPQGL PGLAGTAGEP GRDGNPGSDG LPGRDGSPGG procollagene KGDRGENGSP umano GAPGAPGHPG PPGPVGPAGK SGDRGESGPA GPAGAPGPAG SRGAPGPQGP RGDKGETGER GAAGIKGHRG FPGNPGAPGS PGPAGQQGAI GSPGPAGPRG PVGPSGPPGK 0.2 DGTSGHPGPI GPPGPRGNRG ERGSEGSPGH PGQPGPPGPP GAPGPCCGGV GAAAIAGIGG EKAGGFAPYY GDEPMDFKIN TDEIMTSLKS VNGQIESLIS PDGSRKNPAR NCRDLKFCHP ELKSGEYWVD PNQGCKLDAI KVFCNMETGE TCISANPLNV PRKHWWTDSS AEKKHVWFGE SMDGGFQFSY GNPELPEDVL DVQLAFLRLL 0.1 SSRASQNITY HCKNSIAYMD QASGNVKKAL KLMGSNEGEF KAEGNSKFTY TVLEDGCTKH TGEWSKTVFE YRTRKAVRLP IVDIAPYDIG GPDQEFGVDV GPVCFL Frequenza 0 A C D E F G H I K L M N P Q R S T Y V W aminoacido
27 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento a) Generazione di un ampio numero di sequenze casuali con la stessa composizione della seq query (sequenze shuffled ) b) Ripetizione della ricerca di similarita su sottoinsiemi casuali dei DB utilizzando come query ciascuna delle seq. casuali c) Calcolo degli opt corrispondenti, del loro valore medio M casuale e della corrispondente deviazione standard σ casuale Distribuzione dei punteggi casuali
28 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento Due sequenze possono essere considerate omologhe se il punteggio per il loro allineamento ottimale (opt) cade fuori dalla distribuzione dei punteggi ottenuti per caso Score significativo
29 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento Score di sequenze casuali: distribuzione di Poisson P = 1-exp(-Κmn exp(-λs)) Κ, λ dipendono da matrice e DB m, n lunghezza sequenze comparate M casuale = σ casuale = Σ (opt i ) i n Σ (opt i -M casuale ) i n
30 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento Calcolo dello Z-score e dell E-value per l allineamento della sequenza query con le sue possibili omologhe Z-score = numero di deviazioni standard che separano lo punteggio (opt) della query della media dei punteggi casuali Z-score = (opt query M casuale ) / σ casuale Score significativo Z-score 4 opt query fuori dalla distribuzione casuale.
31 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento E-value = expectation value: numero atteso di sequenze che danno per caso il punteggio S (o opt) Indica quanto e probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio M casuale E-value = Κmn exp(-λs) = mn 2 -S Bit score : S = (λs lnκ) / ln 2 (Ricordarsi che 2 1/ln2 = e) E possibile confrontare direttamente i bit score per ricerche con diverse matrici e diverse banche dati
32 Si tratta di sequenze omologhe? Valutazione della significatività dell allineamento Opt grande Z-score > 4 E-value > 0.01 Bit-score grande

33

34

35
36 BLAST (Basic Local Alignment Search Tool) 1. Suddivide la sequenza query in parole (default, 3 aa) 2. Seleziona parole molto simili mediante matrici di similarità e quindi per ogni tripletta della query genera una famiglia di triplette 3. Mediante matrici di similarità confronta ogni parola con regioni di uguali dimensioni delle entries del DB e ne calcola lo score 4. Se lo score e di un valore soglia T al di sotto del quale la similarita e considerata troppo bassa, estende la regione allineata cercando regioni di alta similarità (score sopra un secondo valore di soglia S), fermandosi quando lo score non può più essere migliorato
37 Sequenza query parole DB Score > T? No elimina Si Identifica la sequenza Sequenze della banca dati Estendi il match di ogni parola finche il punteggio rimane maggiore di s
38 BLAST Step 1 Data una parola di lunghezza w (di solito 3 per le proteine) e una matrice di scoring (es. BLOSUM62): Crea una lista di tutte le parole (w-lettere) cha danno uno score >T se confrontate con le parole di lunghezza w- della query. Sequenza query L N K C K T P Q G Q R L V N Q P Q G 18 P E G 15 P R G 14 P K G 14 P N G 13 P D G 13 P M G 13 Parola della query Parole simili Sotto Soglia (T=13) P Q A 12 P Q N 12 etc.
39 BLAST Step 2 Identifica tutte le posizioni nel database dove si trova una parola sufficientemente simile (hit list). P Q G 18 P E G 15 P R G 14 P K G 14 P N G 13 P D G 13 P M G 13 PMG Database
.")
40 BLAST Step 3 Il programma prova a estendere l allineamento in entrambe le direzioni aggiungendo coppie di residui. I residui sono aggiunti fino a che lo score non è più migliorabile. Considera solo gli allineamenti con score sopra il valore di soglia (S). High Scoring Segment Pairs (Tratto di allineamento ad alto punteggio)
41 BLAST e FASTA differiscono per il modo in cui pescano le potenziali omologhe nel DB (similarità/identità).
42 BLAST e FASTA differiscono per il modo in cui pescano le potenziali omologhe nel DB (similarità/identità). Altra differenza fondamentale tra BLAST e FASTA sta nelle modalita di calcolo della distribuzione dei punteggi casuali: FASTA la calcola ogni volta che gli viene sottomessa una nuova query per la ricerca su un certo DB BLAST usa distribuzioni precalcolate su ciascun DB per insiemi di sequenze casuali di composizione standard E-value = Κmn exp(-λs) n in FASTA = lunghezza della sequenza allineata n in BLAST = lunghezza totale delle sequenze nel DB usato a prescindere dalla query
43 BLAST e FASTA differiscono per il modo in cui pescano le potenziali omologhe nel DB (similarità/identità). Inoltre una differenza fondamentale tra BLAST e FASTA sta nelle modalita di calcolo della distribuzione dei punteggi casuali: FASTA la calcola ogni volta che gli viene sottomessa una nuova query per la ricerca su un certo DB BLAST usa distribuzioni precalcolate su ciascun DB per insiemi di sequenze casuali di composizione standard per questo BLAST maschera le regioni di sequenza della query a bassa complessita
44 BLAST
45 BLAST
46 BLAST

47 BLAST I tratti di sequenza mascherati perche a bassa complessita sono riportati in lettere minuscole colorate in grigio
48 BLAST
49 BLAST

50
51 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
La ricerca di similarità: i metodi
 La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
Z-score. lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random
/ deviazione standard random") Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
Ricerca di omologia di sequenza
 Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
InfoBioLab I ENTREZ. ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche
 InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
Allineamento e similarità di sequenze
 Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Programmazione dinamica
 Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Ricerche con BLAST (Laboratorio)
") Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Le sequenze consenso
 Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Bioinformatica. Analisi del genoma
 Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
Bioinformatica Analisi del genoma GABRIELLA TRUCCO CREMA, 5 APRILE 2017 Cosa è il genoma? Insieme delle informazioni biologiche, depositate nella sequenza di DNA, necessarie alla costruzione e mantenimento
Lezione 6. Analisi di sequenze biologiche e ricerche in database
 Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
BLAST: Basic Local Alignment Search Tool
 BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
Bioinformatica ed applicazioni di bioinformatica strutturale!
 Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini)
") Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Allineamenti di sequenze: concetti e algoritmi
 Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Informatica e biotecnologie II parte
 Informatica e biotecnologie II parte Analisi di sequenze: allineamenti CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Bioinformatica La Bioinformatica è una disciplina
Informatica e biotecnologie II parte Analisi di sequenze: allineamenti CGCTTCGGACGAAATCGCATCAGCATACGATCGCATGCCGGGCGGGATAAC CGAAATCGCATCAGCATACGATCGCATGC Bioinformatica La Bioinformatica è una disciplina
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni:
: Allineamento di sequenze (parte 1) Antonella Meloni:") Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Allineamenti Multipli di Sequenze
 Allineamenti Multipli di Sequenze 1 Allineamento multiplo di sequenze: obiettivi di oggi Definire un allineamento multiplo di sequenze; com è generato; comprendere i principali metodi. Introdurre i database
Allineamenti Multipli di Sequenze 1 Allineamento multiplo di sequenze: obiettivi di oggi Definire un allineamento multiplo di sequenze; com è generato; comprendere i principali metodi. Introdurre i database
Il progetto Genoma Umano è iniziato nel E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA.
 Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Bioinformatics more basic notions
 Bioinformatics more basic notions Alcune slides provengono dal materiale rilasciato da: Dr Sergio Marin Vargas - Verona Prof. Riccardo Percudari - Parma Bioinformatics Bio-inspired Computer science Gli
Bioinformatics more basic notions Alcune slides provengono dal materiale rilasciato da: Dr Sergio Marin Vargas - Verona Prof. Riccardo Percudari - Parma Bioinformatics Bio-inspired Computer science Gli
Lezione 2: Allineamento di sequenze. BLAST e CLUSTALW
 Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Corso di Bioinformatica
 Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
q xi Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p i >0;
 Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Vai al sito: Incolla nel box vuoto la sequenza nucleotidica
 Identificare il gene a cui appartiene la sequenza (sonda) e la sua posizione sul cromosoma. Per raggiungere l obiettivo della prima parte dell attività devi usare il software BLAT (BLAST- Like Alignment
Identificare il gene a cui appartiene la sequenza (sonda) e la sua posizione sul cromosoma. Per raggiungere l obiettivo della prima parte dell attività devi usare il software BLAT (BLAST- Like Alignment
Materiale accessorio al Power Point ALLINEAMENTI DI SEQUENZE_2008. Corso di Bioinformatica per Biotecnologie (G. Colonna).
.") Materiale accessorio al Power Point ALLINEAMENTI DI SEQUENZE_2008. Corso di Bioinformatica per Biotecnologie (G. Colonna). Date due o più sequenze, inizialmente potremmo volere: misurarne il grado di similarità;
Materiale accessorio al Power Point ALLINEAMENTI DI SEQUENZE_2008. Corso di Bioinformatica per Biotecnologie (G. Colonna). Date due o più sequenze, inizialmente potremmo volere: misurarne il grado di similarità;
Omologia di sequenze: allineamento e ricerca
 Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
ESERCITAZIONE 3. OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST
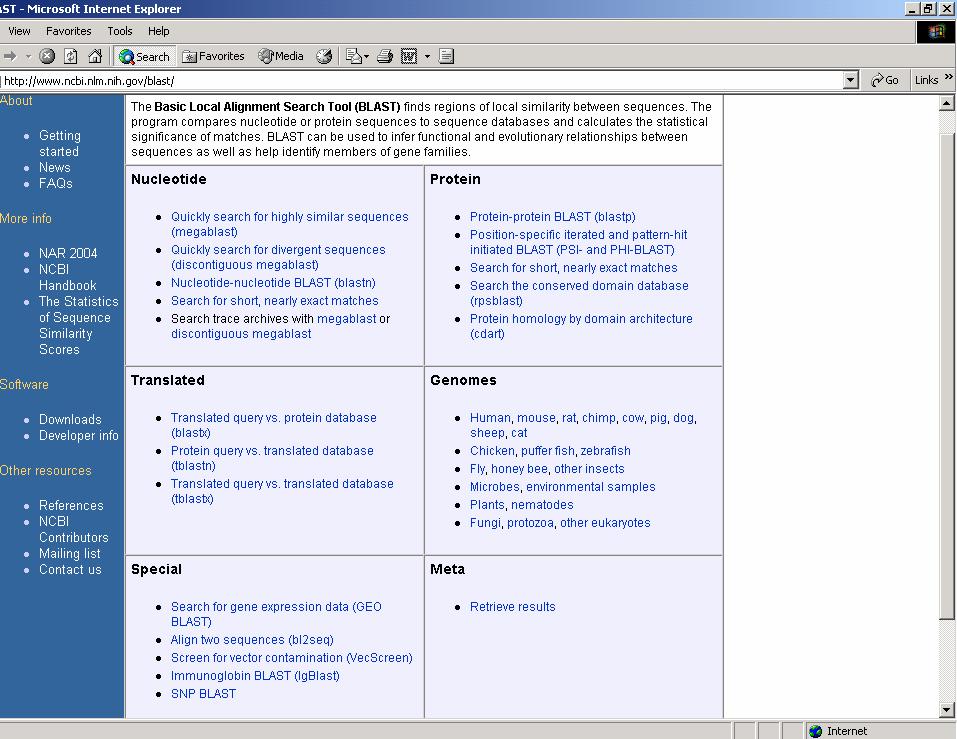
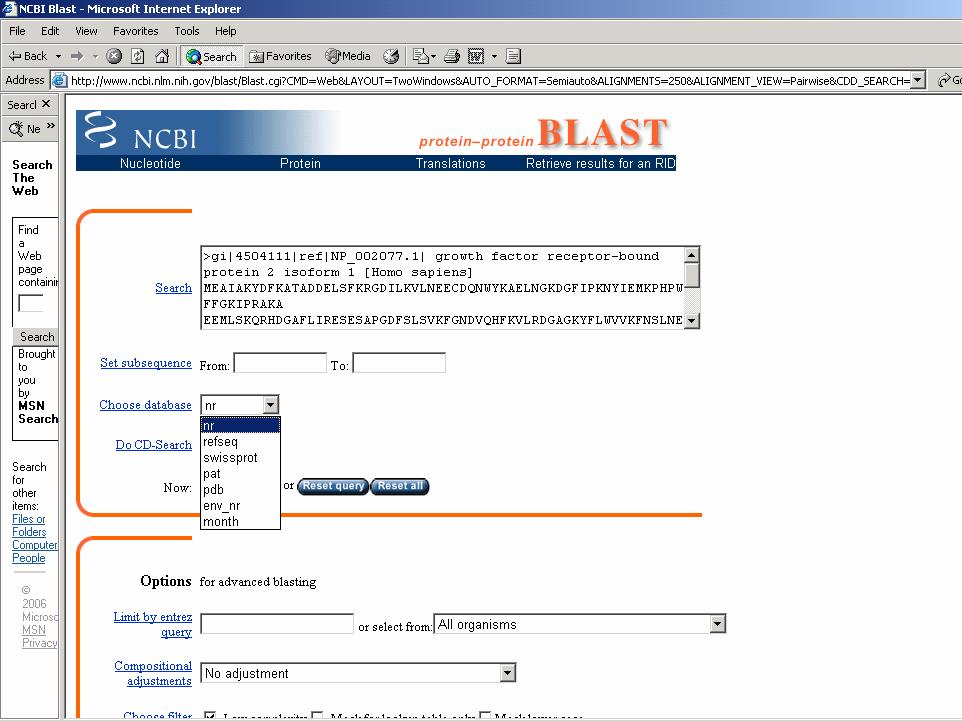
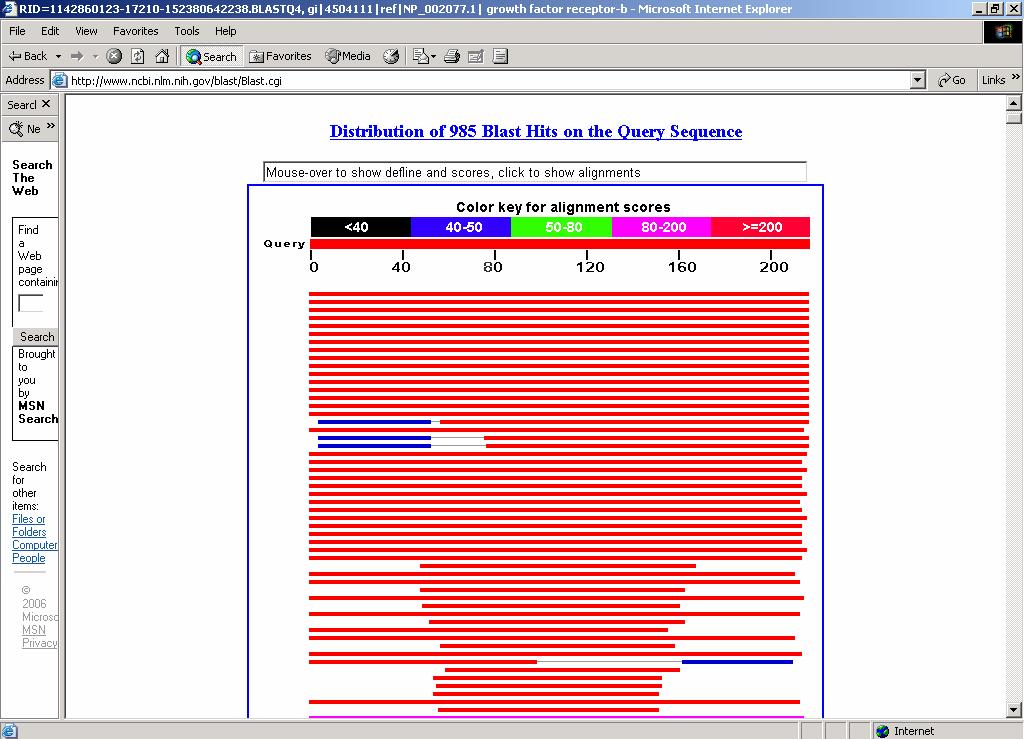
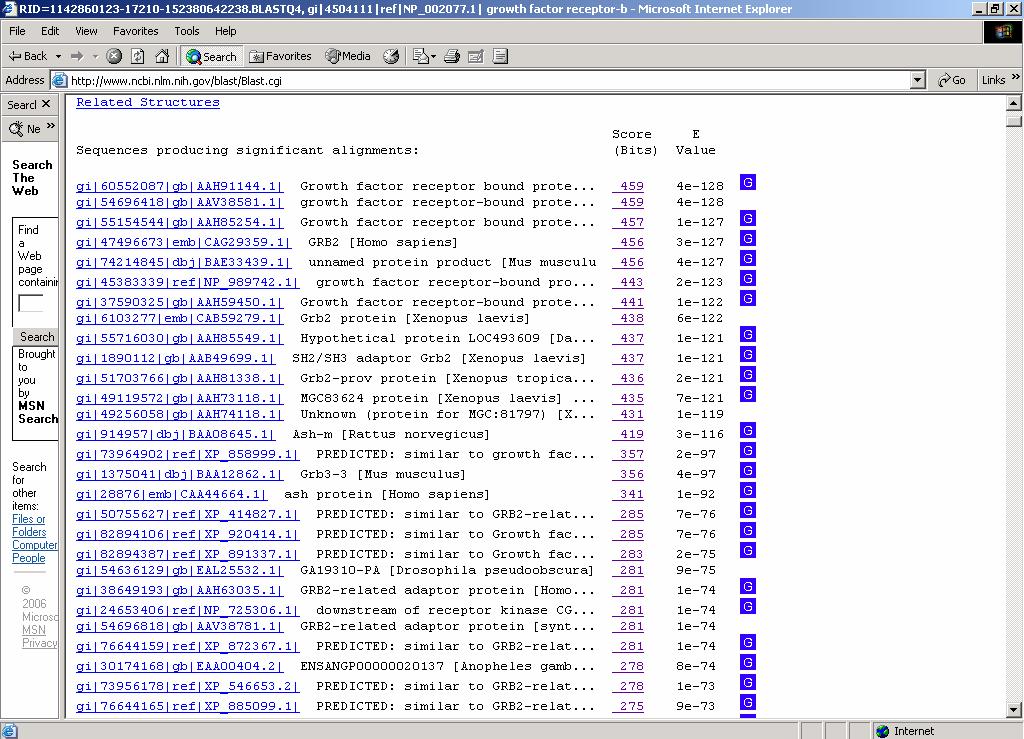
 ESERCITAZIONE 3 OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST L'esercitazione prevede l'utilizzo di risorse web per effettuare ricerche di similarità con la proteina GRB2 (growth factor
ESERCITAZIONE 3 OBIETTIVO: Ricerca di omologhe mediante i programmi FASTA e BLAST L'esercitazione prevede l'utilizzo di risorse web per effettuare ricerche di similarità con la proteina GRB2 (growth factor
Allineamento multiplo
 Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Definizioni
Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Definizioni
Statistica di base per l analisi socio-economica
 Laurea Magistrale in Management e comunicazione d impresa Statistica di base per l analisi socio-economica Giovanni Di Bartolomeo gdibartolomeo@unite.it Definizioni di base Una popolazione è l insieme
Laurea Magistrale in Management e comunicazione d impresa Statistica di base per l analisi socio-economica Giovanni Di Bartolomeo gdibartolomeo@unite.it Definizioni di base Una popolazione è l insieme
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Definizioni preliminari
Riconoscimento e recupero dell informazione per bioinformatica Clustering: similarità Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Definizioni preliminari
Biologia Molecolare Computazionale
 Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
Bioinformatica e Biologia Computazionale per la Medicina Molecolare
 Facoltà di Ingegneria dell Informazione Laurea Specialistica e Magistrale in Ingegneria Informatica Facoltà di Ingegneria dei Sistemi Laurea Magistrale in Ingegneria Biomedica Dipartimento di Elettronica
Facoltà di Ingegneria dell Informazione Laurea Specialistica e Magistrale in Ingegneria Informatica Facoltà di Ingegneria dei Sistemi Laurea Magistrale in Ingegneria Biomedica Dipartimento di Elettronica
RELAZIONE di BIOLOGIA MOLECOLARE
 NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
DNA E PROTEINE IL DNA E RACCHIUSO NEL NUCLEO, MENTRE LA SINTESI PROTEICA SI SVOLGE NEL CITOPLASMA: COME VIENE TRASPORTATA L INFORMAZIONE?
 DNA E PROTEINE NUMEROSI DATI SUGGERISCONO CHE IL DNA SVOLGA IL SUO RUOLO GENETICO CONTROLLANDO LA SINTESI DELLE PROTEINE, IN PARTICOLARE DETERMINANDONE LA SEQUENZA IN AMINOACIDI E NECESSARIO RISPONDERE
DNA E PROTEINE NUMEROSI DATI SUGGERISCONO CHE IL DNA SVOLGA IL SUO RUOLO GENETICO CONTROLLANDO LA SINTESI DELLE PROTEINE, IN PARTICOLARE DETERMINANDONE LA SEQUENZA IN AMINOACIDI E NECESSARIO RISPONDERE
Algoritmi in C++ (seconda parte)
") Algoritmi in C++ (seconda parte) Introduzione Obiettivo: imparare a risolvere problemi analitici con semplici programmi in C++. Nella prima parte abbiamo imparato: generazione di sequenze di numeri casuali
Algoritmi in C++ (seconda parte) Introduzione Obiettivo: imparare a risolvere problemi analitici con semplici programmi in C++. Nella prima parte abbiamo imparato: generazione di sequenze di numeri casuali
A W T V A S A V R T S I A Y T V A A A V R T S I A Y T V A A A V L T S I
 COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Gli obiettivi degli algoritmi di allineamento di sequenze di acidi nucleici o proteine sono molteplici. Possiamo ricordare la ricerca di similarità nelle banche dati, la costruzione
ALLINEAMENTO DI SEQUENZE Gli obiettivi degli algoritmi di allineamento di sequenze di acidi nucleici o proteine sono molteplici. Possiamo ricordare la ricerca di similarità nelle banche dati, la costruzione
CROMOSOMI SESSUALI e ALLELI
 CROMOSOMI SESSUALI e ALLELI Nelle due immagini che seguono possiamo osservare una fotografia dei 46 cromosomi di una cellula somatica umana (a sinistra) e gli stessi cromosomi sistemati, grazie ad un programma
CROMOSOMI SESSUALI e ALLELI Nelle due immagini che seguono possiamo osservare una fotografia dei 46 cromosomi di una cellula somatica umana (a sinistra) e gli stessi cromosomi sistemati, grazie ad un programma
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I 1 INTRODUZIONE
 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 1 INTRODUZIONE Motivazioni dell esistenza della biologia computazionale: Biologia Computazionale
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 1 INTRODUZIONE Motivazioni dell esistenza della biologia computazionale: Biologia Computazionale
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Protein folding. Un gran numero di interazioni deboli + ΔH
 Protein folding -ΔS Un gran numero di interazioni deboli +ΔS + ΔH E r Protein structure modelling: A digression I polimeri (inclusi quelli di amino acidi) in generale non hanno una struttura unica. Le
Protein folding -ΔS Un gran numero di interazioni deboli +ΔS + ΔH E r Protein structure modelling: A digression I polimeri (inclusi quelli di amino acidi) in generale non hanno una struttura unica. Le
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre II. 4 Evoluzione e filogenesi
 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre II 4 Evoluzione e filogenesi FILOGENETICA CS Definzione Studio delle relazioni evolutive tra
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre II 4 Evoluzione e filogenesi FILOGENETICA CS Definzione Studio delle relazioni evolutive tra
Lezione 1. Le molecole di base che costituiscono la vita
 Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
Variabili casuali. - di Massimo Cristallo -
 Università degli Studi di Basilicata Facoltà di Economia Corso di Laurea in Economia Aziendale - a.a. 2012/2013 lezioni di statistica del 16 e 27 maggio 2013 - di Massimo Cristallo - Variabili casuali
Università degli Studi di Basilicata Facoltà di Economia Corso di Laurea in Economia Aziendale - a.a. 2012/2013 lezioni di statistica del 16 e 27 maggio 2013 - di Massimo Cristallo - Variabili casuali
Teoria e tecniche dei test
 Teoria e tecniche dei test Lezione 9 LA STANDARDIZZAZIONE DEI TEST. IL PROCESSO DI TARATURA: IL CAMPIONAMENTO. Costruire delle norme di riferimento per un test comporta delle ipotesi di fondo che è necessario
Teoria e tecniche dei test Lezione 9 LA STANDARDIZZAZIONE DEI TEST. IL PROCESSO DI TARATURA: IL CAMPIONAMENTO. Costruire delle norme di riferimento per un test comporta delle ipotesi di fondo che è necessario
Corso di Genetica -Lezione 8- Cenci
 Corso di Genetica -Lezione 8- Cenci Mappatura mediante ricombinazione corpo nero; fenotipo dominante N(ero)/N(ero) Su quale cromosoma? Y; N/N X w/w; Cy/Sco; Sb/Ser Tutti maschi occhio bianco sono normali
Corso di Genetica -Lezione 8- Cenci Mappatura mediante ricombinazione corpo nero; fenotipo dominante N(ero)/N(ero) Su quale cromosoma? Y; N/N X w/w; Cy/Sco; Sb/Ser Tutti maschi occhio bianco sono normali
Introduzione all analisi di arrays: clustering.
 Statistica per la Ricerca Sperimentale Introduzione all analisi di arrays: clustering. Lezione 2-14 Marzo 2006 Stefano Moretti Dipartimento di Matematica, Università di Genova e Unità di Epidemiologia
Statistica per la Ricerca Sperimentale Introduzione all analisi di arrays: clustering. Lezione 2-14 Marzo 2006 Stefano Moretti Dipartimento di Matematica, Università di Genova e Unità di Epidemiologia
COME È FATTO? Ogni filamento corrisponde ad una catena di nucleotidi
 Il DNA Il DNA è una sostanza che si trova in ogni cellula e contiene tutte le informazioni sulla forma e sulle funzioni di ogni essere vivente: eppure è una molecola incredibilmente semplice. COME È FATTO?
Il DNA Il DNA è una sostanza che si trova in ogni cellula e contiene tutte le informazioni sulla forma e sulle funzioni di ogni essere vivente: eppure è una molecola incredibilmente semplice. COME È FATTO?
Introduzione alla codifica entropica
 Compressione senza perdite Il problema Introduzione alla codifica entropica Abbiamo un alfabeto di simboli A (nota: non è detto che gli elementi di A siano numeri) Sappiamo che il simbolo a A si presenta
Compressione senza perdite Il problema Introduzione alla codifica entropica Abbiamo un alfabeto di simboli A (nota: non è detto che gli elementi di A siano numeri) Sappiamo che il simbolo a A si presenta
Distribuzioni campionarie. Antonello Maruotti
 Distribuzioni campionarie Antonello Maruotti Outline 1 Introduzione 2 Concetti base Si riprendano le considerazioni fatte nella parte di statistica descrittiva. Si vuole studiare una popolazione con riferimento
Distribuzioni campionarie Antonello Maruotti Outline 1 Introduzione 2 Concetti base Si riprendano le considerazioni fatte nella parte di statistica descrittiva. Si vuole studiare una popolazione con riferimento
standardizzazione dei punteggi di un test
 DIAGNOSTICA PSICOLOGICA lezione! Paola Magnano paola.magnano@unikore.it standardizzazione dei punteggi di un test serve a dare significato ai punteggi che una persona ottiene ad un test, confrontando la
DIAGNOSTICA PSICOLOGICA lezione! Paola Magnano paola.magnano@unikore.it standardizzazione dei punteggi di un test serve a dare significato ai punteggi che una persona ottiene ad un test, confrontando la
Tool di allineamento multiplo a confronto
 di allineamento multiplo a confronto Bioinformatica a.a. 2007/08 Andrea Renieri Matteo Tanca Università di Pisa, Dipartimento di Informatica 11 Dicembre 2007 SCHEMA DELLA PRESENTAZIONE 1 INTRODUZIONE Definizione
di allineamento multiplo a confronto Bioinformatica a.a. 2007/08 Andrea Renieri Matteo Tanca Università di Pisa, Dipartimento di Informatica 11 Dicembre 2007 SCHEMA DELLA PRESENTAZIONE 1 INTRODUZIONE Definizione
Sequence alignment... in parallel!!
 Sequence alignment... in parallel!! Diego Puppin Oggi parliamo di... Introduzione: Allineamento di sequenze Algoritmi basati su Programmazione Dinamica Algoritmo Smith-Waterman Micro-parallelismo Parallelismo
Sequence alignment... in parallel!! Diego Puppin Oggi parliamo di... Introduzione: Allineamento di sequenze Algoritmi basati su Programmazione Dinamica Algoritmo Smith-Waterman Micro-parallelismo Parallelismo
Modello computazionale per la predizione di siti di legame per fattori di trascrizione
 Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
Esercitazione del 2/3/2010- Numeri binari e conversione
 Esercitazione del 2/3/2010- Numeri binari e conversione 1. Conversione binario decimale a. 1101 2? 10 1 1 2 Base 2 La posizione della cifra all interno del numero indica il peso della cifra stessa, cioè
Esercitazione del 2/3/2010- Numeri binari e conversione 1. Conversione binario decimale a. 1101 2? 10 1 1 2 Base 2 La posizione della cifra all interno del numero indica il peso della cifra stessa, cioè
Approssimazione normale alla distribuzione binomiale
 Approssimazione normale alla distribuzione binomiale P b (X r) costoso P b (X r) P(X r) per N grande Teorema: Se la variabile casuale X ha una distribuzione binomiale con parametri N e p, allora, per N
Approssimazione normale alla distribuzione binomiale P b (X r) costoso P b (X r) P(X r) per N grande Teorema: Se la variabile casuale X ha una distribuzione binomiale con parametri N e p, allora, per N
Analisi della varianza
 Università degli Studi di Padova Facoltà di Medicina e Chirurgia Facoltà di Medicina e Chirurgia - A.A. 2009-10 Scuole di specializzazione Lezioni comuni Disciplina: Statistica Docente: dott.ssa Egle PERISSINOTTO
Università degli Studi di Padova Facoltà di Medicina e Chirurgia Facoltà di Medicina e Chirurgia - A.A. 2009-10 Scuole di specializzazione Lezioni comuni Disciplina: Statistica Docente: dott.ssa Egle PERISSINOTTO
Unità aritmetica e logica
 Aritmetica del calcolatore Capitolo 9 Unità aritmetica e logica n Esegue le operazioni aritmetiche e logiche n Ogni altra componente nel calcolatore serve questa unità n Gestisce gli interi n Può gestire
Aritmetica del calcolatore Capitolo 9 Unità aritmetica e logica n Esegue le operazioni aritmetiche e logiche n Ogni altra componente nel calcolatore serve questa unità n Gestisce gli interi n Può gestire
Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation
 Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation Dipartimento di Elettronica e Informazione Politecnico di Milano Evolutionary Computation Raggruppa modelli di calcolo
Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation Dipartimento di Elettronica e Informazione Politecnico di Milano Evolutionary Computation Raggruppa modelli di calcolo
SCHEDA DIDATTICA N 7
 FACOLTA DI INGEGNERIA CORSO DI LAUREA IN INGEGNERIA CIVILE CORSO DI IDROLOGIA PROF. PASQUALE VERSACE SCHEDA DIDATTICA N 7 LA DISTRIBUZIONE NORMALE A.A. 01-13 La distribuzione NORMALE Uno dei più importanti
FACOLTA DI INGEGNERIA CORSO DI LAUREA IN INGEGNERIA CIVILE CORSO DI IDROLOGIA PROF. PASQUALE VERSACE SCHEDA DIDATTICA N 7 LA DISTRIBUZIONE NORMALE A.A. 01-13 La distribuzione NORMALE Uno dei più importanti
Statistica Applicata all edilizia: il modello di regressione
 Statistica Applicata all edilizia: il modello di regressione E-mail: orietta.nicolis@unibg.it 27 aprile 2009 Indice Il modello di Regressione Lineare 1 Il modello di Regressione Lineare Analisi di regressione
Statistica Applicata all edilizia: il modello di regressione E-mail: orietta.nicolis@unibg.it 27 aprile 2009 Indice Il modello di Regressione Lineare 1 Il modello di Regressione Lineare Analisi di regressione
Sommario. Presentazione dell opera Ringraziamenti
 Sommario Presentazione dell opera Ringraziamenti XI XII Capitolo 1 Introduzione alla bioinformatica 1 1.1 Cenni introduttivi 1 1.2 Pietre miliari della bioinformatica 2 1.3 Infrastrutture bioinformatiche
Sommario Presentazione dell opera Ringraziamenti XI XII Capitolo 1 Introduzione alla bioinformatica 1 1.1 Cenni introduttivi 1 1.2 Pietre miliari della bioinformatica 2 1.3 Infrastrutture bioinformatiche
Allineamenti multipli
 Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
05. Errore campionario e numerosità campionaria
 Statistica per le ricerche di mercato A.A. 01/13 05. Errore campionario e numerosità campionaria Gli schemi di campionamento condividono lo stesso principio di fondo: rappresentare il più fedelmente possibile,
Statistica per le ricerche di mercato A.A. 01/13 05. Errore campionario e numerosità campionaria Gli schemi di campionamento condividono lo stesso principio di fondo: rappresentare il più fedelmente possibile,
Gasparini Alessandra BIM
 Gasparini Alessandra 592026-BIM Pfam è una banca dati di famiglie proteiche creata dal Wellcome Trust Sanger Institute a partire dal database Pfamseq basato su Uniprot. Le famiglie proteiche di Pfam sono
Gasparini Alessandra 592026-BIM Pfam è una banca dati di famiglie proteiche creata dal Wellcome Trust Sanger Institute a partire dal database Pfamseq basato su Uniprot. Le famiglie proteiche di Pfam sono
Alberi filogenetici. File: alberi_filogenetici.odp. Riccardo Percudani 02/03/04
 Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
A lezione sono stati presentati i seguenti passi per risolvere un problema:
 Calcolo delle radici di un polinomio Problema: Dati i coefficienti a,b,c di un polinomio di 2 grado della forma: ax^2 + bx + c = 0, calcolare le radici. A lezione sono stati presentati i seguenti passi
Calcolo delle radici di un polinomio Problema: Dati i coefficienti a,b,c di un polinomio di 2 grado della forma: ax^2 + bx + c = 0, calcolare le radici. A lezione sono stati presentati i seguenti passi
Esame di Probabilità e Statistica del 30 marzo 2009 (Corso di Laurea Triennale in Matematica, Università degli Studi di Padova).
.") Esame di Probabilità e Statistica del 30 marzo 2009 (Corso di Laurea Triennale in Matematica, Università degli Studi di Padova). Cognome Nome Matricola Es. 1 Es. 2 Es. 3 Es. 4 Somma Voto finale Attenzione:
Esame di Probabilità e Statistica del 30 marzo 2009 (Corso di Laurea Triennale in Matematica, Università degli Studi di Padova). Cognome Nome Matricola Es. 1 Es. 2 Es. 3 Es. 4 Somma Voto finale Attenzione:
Verifica delle ipotesi: Binomiale
 Verifica delle ipotesi: Binomiale Esercizio Nel collegio elettorale di una città, alle ultime elezioni il candidato A ha ottenuto il 4% delle preferenze mentre il candidato B il 6%. Nella nuova tornata
Verifica delle ipotesi: Binomiale Esercizio Nel collegio elettorale di una città, alle ultime elezioni il candidato A ha ottenuto il 4% delle preferenze mentre il candidato B il 6%. Nella nuova tornata
Dal gene alla proteina
 Dal gene alla proteina Il collegamento tra geni e proteine La trascrizione e la traduzione sono i due principali processi che legano il gene alla proteina: uno sguardo panoramico Le informazioni genetiche
Dal gene alla proteina Il collegamento tra geni e proteine La trascrizione e la traduzione sono i due principali processi che legano il gene alla proteina: uno sguardo panoramico Le informazioni genetiche
Introduzione al Calcolo Scientifico A.A Lab. 11
 Introduzione al Calcolo Scientifico A.A. 2009-2010 - Lab. 11 Si consideri il problema dell allineamento di sequenze di proteine in biologia, legato per esempio all annotamento di genomi Si realizzi con
Introduzione al Calcolo Scientifico A.A. 2009-2010 - Lab. 11 Si consideri il problema dell allineamento di sequenze di proteine in biologia, legato per esempio all annotamento di genomi Si realizzi con
Derivazione numerica. Introduzione al calcolo numerico. Derivazione numerica (II) Derivazione numerica (III)
 Derivazione numerica (III)") Derivazione numerica Introduzione al calcolo numerico Il calcolo della derivata di una funzione in un punto implica un processo al limite che può solo essere approssimato da un calcolatore. Supponiamo
Derivazione numerica Introduzione al calcolo numerico Il calcolo della derivata di una funzione in un punto implica un processo al limite che può solo essere approssimato da un calcolatore. Supponiamo
Ricerca di omologhi. La sequenza di cui vogliamo trovare gli omologhi viene de6a query.
 Ricerca di omologhi La sequenza di cui vogliamo trovare gli omologhi viene de6a query. Dobbiamo cercare i suoi omologhi in una banca da= di sequenze (qualche decina di milioni) Allineamento con ciascuna
Ricerca di omologhi La sequenza di cui vogliamo trovare gli omologhi viene de6a query. Dobbiamo cercare i suoi omologhi in una banca da= di sequenze (qualche decina di milioni) Allineamento con ciascuna
Stima dei parametri di modelli lineari
 Stima dei parametri di modelli lineari Indice Introduzione................................ 1 Il caso studio................................ 2 Stima dei parametri............................ 3 Bontà delle
Stima dei parametri di modelli lineari Indice Introduzione................................ 1 Il caso studio................................ 2 Stima dei parametri............................ 3 Bontà delle
Quanti soggetti devono essere selezionati?
 Quanti soggetti devono essere selezionati? Determinare una appropriata numerosità campionaria già in fase di disegno dello studio molto importante è molto Studi basati su campioni troppo piccoli non hanno
Quanti soggetti devono essere selezionati? Determinare una appropriata numerosità campionaria già in fase di disegno dello studio molto importante è molto Studi basati su campioni troppo piccoli non hanno
Esempi di ricerca-dati con Total Materia
 www.totalmateria.com/it Esempi di ricerca-dati con Total Materia 1 Forniamo di seguito esempi applicativi riguardanti Total Materia in Edizione WEB, che rappresenta la più complete e avanzata Banca-Dati
www.totalmateria.com/it Esempi di ricerca-dati con Total Materia 1 Forniamo di seguito esempi applicativi riguardanti Total Materia in Edizione WEB, che rappresenta la più complete e avanzata Banca-Dati
6) Una cellula con 10 coppie di cromosomi entra in mitosi. Quanti cromosomi avrà ognuna delle due cellule figlie? a) 5 b) 20 coppie e) 20 d) 10
 Una cellula con 10 coppie di cromosomi entra in mitosi. Quanti cromosomi avrà ognuna delle due cellule figlie? a) 5 b) 20 coppie e) 20 d) 10") Corso di Laurea in Scienze Ambientali Genetica e Biologia delle popolazioni Autovalutazione n. 1 1)Il fenotipo è: a) l insieme dei geni di un organismo b) il numero di cromosomi di un individuo c) la capacità
Corso di Laurea in Scienze Ambientali Genetica e Biologia delle popolazioni Autovalutazione n. 1 1)Il fenotipo è: a) l insieme dei geni di un organismo b) il numero di cromosomi di un individuo c) la capacità
Calcolo combinatorio
 Fondamenti di Informatica per la Sicurezza a.a. 2007/08 Calcolo combinatorio Stefano Ferrari UNIVERSITÀ DEGLI STUDI DI MILANO DIPARTIMENTO DI TECNOLOGIE DELL INFORMAZIONE Stefano Ferrari Università degli
Fondamenti di Informatica per la Sicurezza a.a. 2007/08 Calcolo combinatorio Stefano Ferrari UNIVERSITÀ DEGLI STUDI DI MILANO DIPARTIMENTO DI TECNOLOGIE DELL INFORMAZIONE Stefano Ferrari Università degli
La codifica digitale
 La codifica digitale Codifica digitale Il computer e il sistema binario Il computer elabora esclusivamente numeri. Ogni immagine, ogni suono, ogni informazione per essere compresa e rielaborata dal calcolatore
La codifica digitale Codifica digitale Il computer e il sistema binario Il computer elabora esclusivamente numeri. Ogni immagine, ogni suono, ogni informazione per essere compresa e rielaborata dal calcolatore
PROBABILITA. Distribuzione di probabilità
 DISTRIBUZIONI di PROBABILITA Distribuzione di probabilità Si definisce distribuzione di probabilità il valore delle probabilità associate a tutti gli eventi possibili connessi ad un certo numero di prove
DISTRIBUZIONI di PROBABILITA Distribuzione di probabilità Si definisce distribuzione di probabilità il valore delle probabilità associate a tutti gli eventi possibili connessi ad un certo numero di prove
Il Codice Gene,co. Il dogma centrale, il flusso dell informazione genica e la decifrazione della informazione del DNA
 Corso di Laurea in Chimica e Tecnologie Farmaceu,che a.a. 2014-2015 Università di Catania Il Codice Gene,co Il dogma centrale, il flusso dell informazione genica e la decifrazione della informazione del
Corso di Laurea in Chimica e Tecnologie Farmaceu,che a.a. 2014-2015 Università di Catania Il Codice Gene,co Il dogma centrale, il flusso dell informazione genica e la decifrazione della informazione del
LA SINTESI PROTEICA LE MOLECOLE CHE INTERVENGONO IN TALE PROCESSO SONO:
 LA SINTESI PROTEICA La sintesi proteica è il processo che porta alla formazione delle proteine utilizzando le informazioni contenute nel DNA. Nelle sue linee fondamentali questo processo è identico in
LA SINTESI PROTEICA La sintesi proteica è il processo che porta alla formazione delle proteine utilizzando le informazioni contenute nel DNA. Nelle sue linee fondamentali questo processo è identico in
Strumenti di indagine per la valutazione psicologica
 Strumenti di indagine per la valutazione psicologica 2.3 Validazione di un test clinico Davide Massidda davide.massidda@gmail.com Definire un cut-off Per ogni scala del questionario, sommando o mediando
Strumenti di indagine per la valutazione psicologica 2.3 Validazione di un test clinico Davide Massidda davide.massidda@gmail.com Definire un cut-off Per ogni scala del questionario, sommando o mediando
TEST DI AUTOVALUTAZIONE APPROSSIMAZIONE NORMALE
 TEST DI AUTOVALUTAZIONE APPROSSIMAZIONE NORMALE I diritti d autore sono riservati. Ogni sfruttamento commerciale non autorizzato sarà perseguito. Metodi statistici per la biologia Parte A. Sia X, X,...
TEST DI AUTOVALUTAZIONE APPROSSIMAZIONE NORMALE I diritti d autore sono riservati. Ogni sfruttamento commerciale non autorizzato sarà perseguito. Metodi statistici per la biologia Parte A. Sia X, X,...
Comunicazioni Elettriche Esercizi
 Comunicazioni Elettriche Esercizi Alberto Perotti 9 giugno 008 Esercizio 1 Un processo casuale Gaussiano caratterizzato dai parametri (µ = 0, σ = 0.5) ha spettro nullo al di fuori dellintervallo f [1.5kHz,
Comunicazioni Elettriche Esercizi Alberto Perotti 9 giugno 008 Esercizio 1 Un processo casuale Gaussiano caratterizzato dai parametri (µ = 0, σ = 0.5) ha spettro nullo al di fuori dellintervallo f [1.5kHz,
II Università degli Studi di Roma
 Versione preliminare gennaio TOR VERGATA II Università degli Studi di Roma Dispense di Geometria. Capitolo 3. 7. Coniche in R. Nel Capitolo I abbiamo visto che gli insiemi di punti P lineare di primo grado
Versione preliminare gennaio TOR VERGATA II Università degli Studi di Roma Dispense di Geometria. Capitolo 3. 7. Coniche in R. Nel Capitolo I abbiamo visto che gli insiemi di punti P lineare di primo grado
Esercitazione: La distribuzione NORMALE
 Esercitazione: La distribuzione NORMALE Uno dei più importanti esempi di distribuzione di probabilità continua è dato dalla distribuzione Normale (curva normale o distribuzione Gaussiana); è una delle
Esercitazione: La distribuzione NORMALE Uno dei più importanti esempi di distribuzione di probabilità continua è dato dalla distribuzione Normale (curva normale o distribuzione Gaussiana); è una delle
Nel DNA e nell RNA i nucleotidi sono legati covalentemente da legami fosfodiesterei.
 DNA e RNA Composizione e proprietà Struttura Analisi 1 STRUTTURA DAGLI ACIDI NUCLEICI Nel DNA e nell RNA i nucleotidi sono legati covalentemente da legami fosfodiesterei. I gruppi fosforici hanno pka vicino
DNA e RNA Composizione e proprietà Struttura Analisi 1 STRUTTURA DAGLI ACIDI NUCLEICI Nel DNA e nell RNA i nucleotidi sono legati covalentemente da legami fosfodiesterei. I gruppi fosforici hanno pka vicino
Esercitazione del 09/03/ Soluzioni
 Esercitazione del 09/03/2006 - Soluzioni. Conversione binario decimale ( Rappresentazione dell Informazione Conversione in e da un numero binario, slide 0) a. 0 2? 0 2 Base 2 Si cominciano a contare le
Esercitazione del 09/03/2006 - Soluzioni. Conversione binario decimale ( Rappresentazione dell Informazione Conversione in e da un numero binario, slide 0) a. 0 2? 0 2 Base 2 Si cominciano a contare le