Allineamenti Multipli di Sequenze
|
|
|
- Mariano Salvi
- 6 anni fa
- Visualizzazioni
Transcript
1 Allineamenti Multipli di Sequenze 1
2 Allineamento multiplo di sequenze: obiettivi di oggi Definire un allineamento multiplo di sequenze; com è generato; comprendere i principali metodi. Introdurre i database di allineamenti multipli di sequenze Capire come creare il proprio allineamento multiplo di sequenza Mostrare come un allineamento multiplo di sequenze fornisca la base per alberi filogenetici
3 Allineamento multiplo di sequenze: schema della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking
4 Allineamento multiplo di sequenze: una definizione un allineamento multiplo e una collezione di tre o più sequenze proteiche (o nucleotidiche) parzialmente o completamente allineate I residui e le zone omologhe sono allineate in colonne per tutta la lunghezza delle sequenze Il senso dell omologia dei residui è evoluzionistico Il senso dell omologia dei residui è strutturale Si tratta di un argomento di ricerca attivo dagli anni 90
5 Allineamento multiplo di sequenze: alcuni fatti Non c è necessariamente un allineamento "corretto" per una famiglia di proteine Le sequenze di proteine evolvono Le corrispondenti strutture tridimensionali evolvono, anche se più lentamente Può essere impossibile identificare i residui di aminoacidi che si allineano correttamente (strutturalmente) lungo ogni parte di un allineamento multiplo di sequenze. Due proteine che condividono identità per il 30% degli aminoacidi, avranno circa il 50% dei residui sovrapponibili nelle due strutture
6 Allineamento multiplo di sequenze: caratteristiche utili per la realizzazione Alcuni residui allineati, come cisteine che formano ponti disolfuro, o i triptofani, possono essere altamente conservati Ci possono essere motivi conservati come un dominio transmembrana Alcune caratteristiche come le strutture secondarie, siti attivi e di legame per ligandi o complessi sono significative Ci possono essere regioni con inserimenti o delezioni propagati in parte della famiglia. I principi che vedremo sono focalizzati sulle proteine ma sono validi in generale anche per sequenze nucleotidiche.
7 Allineamento di sequenze multiple: utilizzi e vantaggi Il MSA è più sensibile di quello a coppie nel rilevamento di omologie, per questo e uno strumento essenziale nella costruzione di modelli strutturali per omologia L output di BLAST può assumere la forma di un MSA, e possono essere individuati residui conservati o motivi In un MSA si possono analizzare i dati di una popolazione Una singola query può essere cercata contro un database di MSA (ad esempio Pfam) Le regioni regolatorie dei geni sono spesso identificabili da MSA
8 Allineamento multiplo di sequenze: riepilogo della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking Argomento che non vedremo, non ci sono soluzioni efficienti e già con 5 sequenze il tempo impiegato è eccessivo (esponenziale) Metodi euristici
9 Allineamento multiplo di sequenze: Metodi metodi progressivi: usano un albero guida (analogo ad un albero filogenetico) per determinare come combinare uno per uno allineamenti a coppie (progressivamente) per creare un allineamento multiplo. Esempi: CLUSTALW, MUSCLE (usato da HomoloGene)

10 Usa ClustalW per fare un MSA progressivo
11 Usa ClustalW per fare un MSA progressivo



12 Il concetto alla base di CLUSTALW Nota: tutte le distanze tra la I e la IV riga sono minori di quelle riportate nella V
13 Il MSA progressivo di Feng-Doolittle (1987) alla base di ClustalW avviene in 3 fasi 1. Realizzare una serie di allineamenti a coppie globali (Needleman e Wunsch, algoritmo di programmazione dinamica) di cui si calcola la distanza (matrice delle distanze) 2. Creare un albero guida a partire dalla matrice delle distanze 3. Allineare progressivamente le sequenze
14 MSA progressivo, fase 1 di 3: generare allineamenti a coppie globali Esempio: allineare 5 globine (1, 2, 3, 4, 5). Primo step: a due a due e valutare gli score di ogni possibile allineamento a coppie migliore punteggio (% identità)
15 Numero di allineamenti a coppie necessari per coprire tutte le possibili combinazioni Per n sequenze, (n-1) (n) / 2 Per 5 sequenze, (4) (5) / 2 = 10 Per 200 sequenze, (199) (200) / 2 = Quindi per molte sequenze ClustalW è molto lento ed è meglio preferire metodi più veloci (MUSCLE è molto veloce)
16 Feng-Doolittle fase 2: albero guida Convertire i punteggi di somiglianza in punteggi di distanza: è matematicamente più semplice, oltre che più intuitivo, lavorare con le distanze. Una semplice definizione di distanza possibile e data dalla percentuale di residui diversi (100- SI in %) che viene messa nella matrice delle distanze In ogni casella c e la % di residui diversi (o altra misura di distanza) tra le due sequenze
17 Feng-Doolittle fase 2: albero guida Dalla matrice delle distanze si calcola l albero guida con il metodo di clustering neighbor joining che discuteremo poi Tutte le sequenze vengono poi allineate progressivamente, seguendo le indicazioni dell albero guida: prima si allineano le piu simili (vicine) e poi progressivamente le piu distanti A ogni passaggio si utilizza un algoritmo dinamico di allineamento molto efficiente che accoppia sequenze o gruppi di sequenze Il MA e composto a partire a tanti allineamenti a coppie, anche fra gruppi di sequenze. NOTA: Le indel presenti negli allineamenti gia effettuati restano fisse.
18 Feng-Doolittle fase 3: allineamento progressivo Fare un MSA in base all'ordine nell albero guida: Iniziare con le due sequenze più strettamente correlate Quindi aggiungere la sequenza (o il gruppo) successiva più vicina Continuare fino a quando tutte le sequenze vengono aggiunte al MSA Regola: Un gap è per sempre
19 Come allineare pogressivamente due gruppi di sequenze? Si usa sempre una matrice. E simile all allineamento dinamico di due sequenze visto. Lo score S in ogni casella e la media degli score ottenuti confrontando tutte le possibili coppie di a.a. nella riga e colonna corrispondenti (secondo ad es. BLOSUM62) Quando la matrice e stata totalmente calcolata, si calcola il percorso con il miglior allineamento (unisce gli S piu alti)
20 Perché un Gap è per sempre"? Ci sono molti modi possibili per fare un MSA Dove aggiungere i gaps è una questione cruciale: i gaps sono spesso aggiunti tra le prime due sequenze (le più vicine) Cambiare la scelta iniziale di un gap successivamente significherebbe dare più peso, nella definizione dell evoluzione della famiglia a sequenze più distanti tra loro. Mantenere le scelte dei gap iniziale significa credere che quell evento sia più affidabile, quindi se serve un gap, prima si valuta dove ne sono già stati visti e poi si considerano altre posizioni.
21 Allineamento multiplo di sequenze: Metodi Esempio di MSA con ClustalW: due insiemi di dati Cinque globine lontanamente correlate (umano a pianta) Cinque globine beta strettamente correlate (specie differenti)
22 ClustalW allineamento di 5 globine distanti Output E un allineamento globale. * :. e spazio nell ordine, indicano la bontà dell appaiamento. * = identità

23 ClustalW allineamento di 5 globine vicine Output E un allineamento globale. * :. e spazio nell ordine, indicano la bontà dell appaiamento. * = identità
24 ClustalW: come migliorare la sua capacità di generare MSA accurati Alle sequenze allineate vengono dati dei pesi. Gruppi di sequenze strettamente correlate hanno un peso minore se nell allineamento ve ne sono altre più distanti, per evitare che le più simili dominino l allineamento. Diverse matrici di punteggio e penalizzazione degli indel vengono usate durante l allineamento in base alla presenza di sequenze conservate o divergenti da allineare in quello step, ad esempio: PAM20 PAM60 PAM120 PAM % id 60-80% id 40-60% id 0-40% id Sanzioni residuo-specifiche sono applicate per i gap, per indurre la concentrazione in posizioni in cui già ci sono.
 per una spiegazione dettagliata delle tre tappe dell")
25 Vedi Thompson et al. (1994) per una spiegazione dettagliata delle tre tappe dell allineamento progressivo attuato in ClustalW
26 Matrice di tutte le possibili distanze (equivalente alla similitudine) tra le coppie 1) Allineamento a coppie: calcola la matrice delle distanze 2) Albero (senza radice) che correla i vicini
27 2) Albero (senza radice) che correla i vicini 2) Albero (con radice) che correla i vicini e con i pesi per le sequenze (in base a distanze e altri parametri): Albero Guida Pesi per l allineamento multiplo

28 2) Albero (con radice) che correla i vicini e con i pesi per le sequenze (in base a distanze e altri parametri): Albero Guida 3) Allineamento progressivo: allinea seguendo l albero guida. Tra gruppi di 2 o più sequenze i confronti vengono fatti tra profili.
29 Esempio sulla variabilità dei MSA: allineamenti di 5 globine utilizzando 5 diversi programmi Consideriamo un allineamento multiplo di sequenze (MSA) di cinque globine. Useremo cinque tra i migliori programmi per il MSA: ClustalW, Praline, MUSCLE (utilizzato da HomoloGene), ProbCons e TCoffee. Ogni programma offre punti di forza particolari. Ci concentreremo su una istidina (H) residuo che ha un ruolo critico nel legare l'ossigeno nelle globine, e dovrebbe quindi essere allineata; tuttavia spesso non è allineata. Ciascuno dei cinque programmi può dare risposte diverse. La nostra conclusione è che non esiste un approccio ottimale al MSA. Decine di nuovi programmi sono stati introdotti negli ultimi anni.
 vari a seconda di quale dei cinque algoritmi è utilizzato H allineata meno bene.")
30 ClustalW Il più usato, è uscito nel 1994 e non viene piu agiornato. E anche quello di riferimento per i nuovi programmi che spesso hanno prestazioni migliori. H ben conservata e allineata Nota come la regione in cui una istidina è conservata ( ) vari a seconda di quale dei cinque algoritmi è utilizzato H allineata meno bene. Regione più difficile da allineare
 vari a seconda di quale dei cinque algoritmi è utilizzato Qui l allineamento dell H non è")
31 Praline Introdotto di recente, ha buone prestazioni Punteggio di affidabilità Nota come la regione in cui una istidina è conservata ( ) vari a seconda di quale dei cinque algoritmi è utilizzato Qui l allineamento dell H non è buono

32 MUSCLE Nuovo programma (molto buono e molto veloce) che sta avendo più successo di ClustalW Qui l allineamento dell H non è buono

33 Probcons Usa le HMM e con MUSCLE sta guadagnando consensi fra gli utilizzatori

34 TCoffee Incorpora informazioni sulla struttura secondaria nel realizzare l allineamento. Qui l allineamento dell H è corretto
35 Allineamento multiplo di sequenze: schema della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking
36 Allineamento multiplo di sequenze: metodi iterativi Metodi iterativi: consistono nel calcolare una soluzione sub-ottimale e modificarla ripetutamente con metodi della programmazione dinamica o altri metodi fino a quando la soluzione converge (non migliora ulteriormente) Esempi: MAFFT, MUSCLE, IterAlign, Praline Non li vediamo nel dettaglio nel corso

37 Accesso a MUSCLE presso EBI
38 Allineamento multiplo di sequenze: riepilogo della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking
39 Allineamento multiplo di sequenze: la consistenza (T-COFFEE, PRRP, DIALIGN, ProbCons) Algoritmi potenti, veloci e accurati basati sulla consistenza: valutano la probabilità di allineamento sulla base di database di allineamenti a coppie locali ad alto punteggio e allineamenti a coppie globali a lungo raggio per creare un allineamento completo. Concetto: se abbiamo 3 sequenze A, B e C, e allineiamo A con B e B con C, implicitamente abbiamo allineato anche A con C. Ma magari l allineamento implicito è incoerente (o incosistente) rispetto al diretto allineamento A con C: cerco un MSA che ottimizzi la consistenza
40 Approccio basato sulla consistenza: ProbCons Sequenza x Sequenza y Sequenza z x i y j z k Se x i si allinea con z k e z k si allinea con y j allora x i dovrebbe allinearsi con y j. Il programma determina vincoli che cercano di soddisfare questa necessita di coerenza. ProbCons incorpora elementi di prova da più sequenze per guidare la creazione di un allineamento a coppie. E accessibile presso

41 Output di ProbCons: le iterazioni sulla consistenza aiutano
42 Allineamento multiplo di sequenze: schema della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking


43 Accesso a TCoffee: Una collezione di tools per MSA Fa un MSA sulla consistenza Confronta 15 metodi di MSA Fa un MSA con RNA MSA con dati strutturali MSA con dati di omologia Combina diversi metodi di MSA Valuta MSA in base alla consistenza Valuta MSA in base alla struttura
44 TCoffee può integrare le informazioni strutturali in un MSA Qualità dell allineamento in relazione alle strutture Protein Data Bank accession numbers
45 Allineamento multiplo di sequenze: riepilogo della lezione 1. Introduzione al MSA Metodi esatti Progressivi (ClustalW) Iterativi (MUSCLE) Basati sulla consistenza (ProbCons) Basati sulla struttura (Expresso) Conclusioni: studi di benchmarking
46 Allineamento multiplo di sequenze: Metodi Come facciamo a decidere quale programma utilizzare? Ci sono dataset di benchmarking per gli allineamenti multipli allineati meticolosamente a mano, in base a somiglianza strutturale, o con algoritmi automatici ma molto accurati (dispendiosi in termini di tempo e memoria). Alcuni programmi dotati di interfacce che sono più userfriendly rispetto ad altri. La maggior parte dei programmi sono molto buoni, dunque dipende dalla vostra preferenza. Se le vostre proteine hanno una struttura 3D, usatela per aiutarvi a giudicare l allineamento. Ad esempio, provare con Expresso.
47 Strategia per la valutazione degli algoritmi per l allineamento multiplo (benchmarking) 1) Creare o utilizzare un database di sequenze proteiche per le quali la struttura 3D è nota. Così possiamo definire i "veri" omologhi in base ai criteri strutturali. 2) Prova a fare allineamenti multipli di sequenza con molti gruppi diversi di proteine (molto correlati, molto lontani, pochi gap, molti gap, inserzioni, outliers). 3) Confrontare le risposte e scegliere il migliore algoritmo per quel set
48 BAliBASE: A benchmark alignments database for the evaluation of multiple sequence alignment programs (database di allineamenti affidabili)
49 Strategia per la valutazione degli algoritmi per l allineamento multiplo (benchmarking) IN CONCLUSIONE: Test di valutazione suggeriscono che ProbCons (algoritmo basato sulla consistenza/progressivo), dà i migliori risultati con BAliBASE, anche se MUSCLE (algoritmo progressivo) è un programma estremamente veloce e preciso. ClustalW è il programma più usato. Ha una bella interfaccia (in particolare con ClustalX) ed è facile da usare. Ma diversi programmi sono migliori. Non c'è IL programma migliore: gli output possono essere certamente diversi (soprattutto se si allineare proteine divergenti o sequenze di DNA)
50 Il database Pfam (famiglie di proteine) è una risorsa importante per l'analisi delle famiglie di proteine

51 Database Pfam (famiglia di proteine) grafico della conservazione dei residui

52 Pfam- il visualizzatore JalView
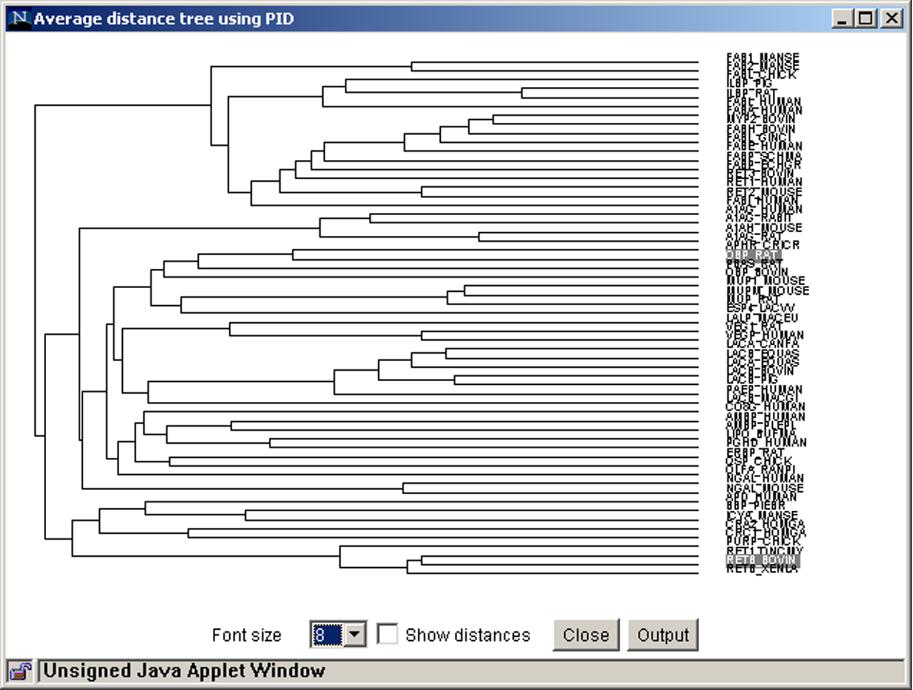
53

54 Rielaborazione e visualizzazione di MSA Esistono vari tools utili per visualizzare e rielaborare gli allineamenti multipli: es. Jalview (

55 Interfacciato a predittori di struttura secondaria.


56 Interfacciato a UNIPROT. E molti altri tools pratici (costruzione di alberi, visualizzazione di strutture ecc.)

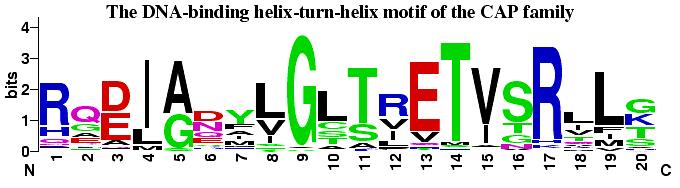
57 WebLogo (
58 Cenno: allineamento multiplo di sequenze di DNA genomico L obiettivo è quello di distinguere regioni più dinamiche da quelle più conservate. Negli allineamenti di DNA genomico ci sono in genere poche sequenze (fino a diverse decine), ognuno anche di milioni di paia di basi. L aggiunta di più specie migliora la precisione. L'allineamento delle sequenze divergenti rivela spesso isole di conservazione (producendo "ancore" per l'allineamento). I cromosomi sono soggetti a inversioni, duplicazioni, delezioni e traslocazioni (che spesso coinvolgono milioni di paia di basi). Ad esempio il cromosoma 2 umano deriva dalla fusione di due cromosomi acrocentrici. Non ci sono set di dati di riferimento disponibili.
 Fase 1: in NCBI cambiare il menu a tendina per HomoloGene e immettere caveolina nella casella di")
59 Un modo rapido per accedere ad allineamenti multipli: Homologene La caveolina (proteina presente nelle caveole della membrana plasmatica, raft lipidici ricchi di colesterolo e sfingolipidi) Fase 1: in NCBI cambiare il menu a tendina per HomoloGene e immettere caveolina nella casella di ricerca

60 Fase 2: verificare i risultati. Prendiamo la prima serie di caveolin 3. Visualizzare l allineamento multiplo. Allineamenti multipli precalcolati tra omologhi

61 Fase 3: controllare l'allineamento multiplo. Si noti che questi otto proteine si allineano bene, anche se devono essere inclusi alcuni gaps. Gap terminale Gap interno Inserimento o delezione? Come va assegnato il punteggio? RefSeq

62 Ecco un altro allineamento multiplo, Rac: Questo inserimento può essere a causa di splicing alternativo
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Le sequenze consenso
 Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
Le sequenze consenso Si definisce sequenza consenso una sequenza derivata da un multiallineamento che presenta solo i residui più conservati per ogni posizione riassume un multiallineamento. non è identica
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
InfoBioLab I ENTREZ. ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche
 InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
ALLINEAMENTI MULTIPLI
 ALLINEAMENTI MULTIPLI Identificazione di siti funzionalmente importanti Dimostrazione di omologia Filogenesi molecolare Ricerca di somiglianze deboli ma significative in banche dati Predizione di struttura
ALLINEAMENTI MULTIPLI Identificazione di siti funzionalmente importanti Dimostrazione di omologia Filogenesi molecolare Ricerca di somiglianze deboli ma significative in banche dati Predizione di struttura
Allineamento multiplo
 Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Allineamento multiplo Allineamenti multipli Il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione analoga. I siti funzionalmente o strutturalmente
Allineamenti di sequenze: concetti e algoritmi
 Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
q xi Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p i >0;
 Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini)
") Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
Esercizio: Ricerca di sequenze in banche dati e allineamento multiplo (adattato da una lezione del Prof. Paiardini) Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
Relazione sequenza-struttura e funzione
 Biotecnologie applicate alla progettazione e sviluppo di molecole biologicamente attive A.A. 2010-2011 Modulo di Biologia Strutturale Relazione sequenza-struttura e funzione Marco Nardini Dipartimento
Biotecnologie applicate alla progettazione e sviluppo di molecole biologicamente attive A.A. 2010-2011 Modulo di Biologia Strutturale Relazione sequenza-struttura e funzione Marco Nardini Dipartimento
Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Laboratorio di Bioinformatica I. Filogenesi. Dott. Sergio Marin Vargas (2014 / 2015)
") Laboratorio di Bioinformatica I Filogenesi Dott. Sergio Marin Vargas (2014 / 2015) Evoluzione Selezione Naturale Selezione Artificiale Variazione casuale Risultato Variazioni Casuali Mutazioni favorite
Laboratorio di Bioinformatica I Filogenesi Dott. Sergio Marin Vargas (2014 / 2015) Evoluzione Selezione Naturale Selezione Artificiale Variazione casuale Risultato Variazioni Casuali Mutazioni favorite
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
Ricerche con BLAST (Laboratorio)
") Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Laboratorio di Bioinformatica I Ricerche con BLAST (Laboratorio) Dott. Sergio Marin Vargas (2014 / 2015) NCBI BLAST BLAST: Basic Local Alignment Search Tool http://blast.ncbi.nlm.nih.gov/blast.cgi NCBI
Laboratorio di Algoritmi e Strutture Dati
 Laboratorio di Algoritmi e Strutture Dati Docente: Camillo Fiorentini 8 gennaio 8 Il problema è simile all esercizio 5.6 del libro di testo di algoritmi (Introduzione agli algoritmi e strutture dati, T.
Laboratorio di Algoritmi e Strutture Dati Docente: Camillo Fiorentini 8 gennaio 8 Il problema è simile all esercizio 5.6 del libro di testo di algoritmi (Introduzione agli algoritmi e strutture dati, T.
8. Sovrapposizione e confronto di strutture proteiche
 8. Sovrapposizione e confronto di strutture proteiche In questa esercitazione utilizzeremo il programma SwissPdbViewer (versione 4.0.1) per effettuare la sovrapposizione e il confronto di strutture proteiche.
8. Sovrapposizione e confronto di strutture proteiche In questa esercitazione utilizzeremo il programma SwissPdbViewer (versione 4.0.1) per effettuare la sovrapposizione e il confronto di strutture proteiche.
Bioinformatica ed applicazioni di bioinformatica strutturale!
 Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Bioinformatica ed applicazioni di bioinformatica strutturale! Bioinformatica! Le banche dati! Programmi per estrarre ed analizzare i dati! I numeri! Cellule nell uomo! Geni nell uomo! Genoma umano Il dogma
Il progetto Genoma Umano è iniziato nel E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA.
 Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
Il progetto Genoma Umano è iniziato nel 1990. E stato possibile perchè nel 1986 era stato sviluppato il sequenziamento automatizzato del DNA. Progetto internazionale finanziato da vari paesi, affidato
BLAST: Basic Local Alignment Search Tool
 BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
BLAST: Basic Local Alignment Search Tool 1 Outline della lezione di oggi BLAST Uso pratico Algoritmo Strategie Trovare proteine lontanamente legate: PSI-BLAST 2 Problema con gli algoritmi dinamici Gli
Lezione 2: Allineamento di sequenze. BLAST e CLUSTALW
 Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Lezione 2: Allineamento di sequenze BLAST e CLUSTALW Allineamento di sequenze Allineamenti L avvento della genomica moderna permette di analizzare le similitudini e le differenze tra organismi a livello
Biologia Molecolare Computazionale
 Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
Biologia Molecolare Computazionale Paolo Provero - paolo.provero@unito.it 2008-2009 Argomenti Allineamento di sequenze Ricostruzione di alberi filogenetici Gene prediction Allineamento Allineamento di
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
GENOMA. Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine CONTENUTO FUNZIONE. Progetti genoma in centinaia di organismi
 GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
GENOMA EVOLUZIONE CONTENUTO FUNZIONE STRUTTURA Analisi di sequenze -- Analisi di espressione -- Funzione delle proteine Progetti genoma in centinaia di organismi Importante la sintenia tra i genomi The
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
Riconoscimento e recupero dell informazione per bioinformatica Filogenesi Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Sommario Introduzione alla
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Allineamenti multipli
 Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte proteine a funzione
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
Ricerca di omologia di sequenza
 Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Z-score. lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random
/ deviazione standard random") Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Z-score lo Z-score è definito come: Z-score = (opt query - M random)/ deviazione standard random è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di quante dev.
Metodo di Quine- McCluskey
 Metodo di Quine- McCluskey Maurizio Palesi Maurizio Palesi Definizioni Date due funzioni f(x,x 2,,x n ) e g(x,x 2,,x n ) si dice che f copre g (oppure g implica f) e si scrive f g se f(x,x 2,,x n )= quando
Metodo di Quine- McCluskey Maurizio Palesi Maurizio Palesi Definizioni Date due funzioni f(x,x 2,,x n ) e g(x,x 2,,x n ) si dice che f copre g (oppure g implica f) e si scrive f g se f(x,x 2,,x n )= quando
Sistemi di Elaborazione delle Informazioni
 SCUOLA DI MEDICINA E CHIRURGIA Università degli Studi di Napoli Federico II Corso di Sistemi di Elaborazione delle Informazioni Dott. Francesco Rossi a.a. 2016/2017 1 I linguaggi di programmazione e gli
SCUOLA DI MEDICINA E CHIRURGIA Università degli Studi di Napoli Federico II Corso di Sistemi di Elaborazione delle Informazioni Dott. Francesco Rossi a.a. 2016/2017 1 I linguaggi di programmazione e gli
Modello computazionale per la predizione di siti di legame per fattori di trascrizione
 Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
Modello computazionale per la predizione di siti di legame per fattori di trascrizione Attività di tirocinio svolto presso il Telethon Institute of Genetics and Medicine Relatori Prof. Giuseppe Trautteur
scaricato da I peptidi risultano dall unione di due o più aminoacidi mediante un legame COVALENTE
 Legame peptidico I peptidi risultano dall unione di due o più aminoacidi mediante un legame COVALENTE tra il gruppo amminico di un aminoacido ed il gruppo carbossilico di un altro. 1 Catene contenenti
Legame peptidico I peptidi risultano dall unione di due o più aminoacidi mediante un legame COVALENTE tra il gruppo amminico di un aminoacido ed il gruppo carbossilico di un altro. 1 Catene contenenti
Alberi filogenetici. File: alberi_filogenetici.odp. Riccardo Percudani 02/03/04
 Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
TASSONOMIA O SISTEMATICA
 TASSONOMIA O SISTEMATICA È la branca della batteriologia responsabile della caratterizzazione degli organismi ed organizzazione in gruppi affini (TAXA). NOMENCLATURA CLASSIFICAZIONE IDENTIFICAZIONE taxon
TASSONOMIA O SISTEMATICA È la branca della batteriologia responsabile della caratterizzazione degli organismi ed organizzazione in gruppi affini (TAXA). NOMENCLATURA CLASSIFICAZIONE IDENTIFICAZIONE taxon
4. Ricerca di sequenze in banche dati e allineamento multiplo
 4. Ricerca di sequenze in banche dati e allineamento multiplo Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni di BLAST disponibili sono organizzate in base al
4. Ricerca di sequenze in banche dati e allineamento multiplo Collegatevi al sito www.ncbi.nlm.nih.gov/blast. Apparirà una pagina nella quale le versioni di BLAST disponibili sono organizzate in base al
BLOWFISH. Introduzione Algoritmo. Prestazioni Cryptanalysis of Vaundenay. Egizio Raffaele
 Introduzione Algoritmo Prestazioni Cryptanalysis of Vaundenay Egizio Raffaele Introduzione E un cifrario a blocchi a chiave simmetrica Utilizza varie tecniche tra le quali la rete Feistel, le S-box dipendenti
Introduzione Algoritmo Prestazioni Cryptanalysis of Vaundenay Egizio Raffaele Introduzione E un cifrario a blocchi a chiave simmetrica Utilizza varie tecniche tra le quali la rete Feistel, le S-box dipendenti
Omologia di sequenze: allineamento e ricerca
 Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Allineamento e similarità di sequenze
 Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
L efficienza e la valutazione delle performance Concetti ed introduzione alla D.E.A.
 L efficienza e la valutazione delle performance Concetti ed introduzione alla D.E.A. Corso di Economia Industriale Lezione dell 8/01/2010 Valutazione delle peformance Obiettivo: valutare le attività di
L efficienza e la valutazione delle performance Concetti ed introduzione alla D.E.A. Corso di Economia Industriale Lezione dell 8/01/2010 Valutazione delle peformance Obiettivo: valutare le attività di
Corso di Bioinformatica
 Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
Corso di Bioinformatica Cortona - Novembre 2002 Metodi Computazionali per l'analisi delle sequenze Dr. Sabino Liuni Istituto di Tecnologie Biomediche- CNR Sezione di Bioinformatica e Genomica - Bari Sabino@area.ba
CALCOLO NUMERICO. Prof. Di Capua Giuseppe. Appunti di Informatica - Prof. Di Capua 1
 CALCOLO NUMERICO Prof. Di Capua Giuseppe Appunti di Informatica - Prof. Di Capua 1 INTRODUZIONE Quando algoritmi algebrici non determinano la soluzione di un problema o il loro «costo» è molto alto, allora
CALCOLO NUMERICO Prof. Di Capua Giuseppe Appunti di Informatica - Prof. Di Capua 1 INTRODUZIONE Quando algoritmi algebrici non determinano la soluzione di un problema o il loro «costo» è molto alto, allora
Allineamenti multipli
 Allineamenti multipli Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte
Allineamenti multipli Allineamenti multipli Finora ci siamo occupati di allineamenti a coppie (pairwise), ma il modo migliore per conoscere le caratteristiche di una determinata famiglia è allineare molte
Modelli di recupero. Modello di recupero booleano
 Modelli di recupero L obiettivo è recuperare i documenti che sono verosimilmente rilevanti all interrogazione. Vi sono vari modelli di recupero, che possono essere suddivisi in due grandi famiglie: exact
Modelli di recupero L obiettivo è recuperare i documenti che sono verosimilmente rilevanti all interrogazione. Vi sono vari modelli di recupero, che possono essere suddivisi in due grandi famiglie: exact
Clustering con Weka. L interfaccia. Prof. Matteo Golfarelli Alma Mater Studiorum - Università di Bologna. Algoritmo utilizzato per il clustering
 Clustering con Weka Soluzioni degli esercizi Prof. Matteo Golfarelli Alma Mater Studiorum - Università di Bologna L interfaccia Algoritmo utilizzato per il clustering E possibile escludere un sottoinsieme
Clustering con Weka Soluzioni degli esercizi Prof. Matteo Golfarelli Alma Mater Studiorum - Università di Bologna L interfaccia Algoritmo utilizzato per il clustering E possibile escludere un sottoinsieme
Fondamenti di Informatica
 Fondamenti di Informatica AlgoBuild: Strutture selettive, iterative ed array Prof. Arcangelo Castiglione A.A. 2016/17 AlgoBuild : Strutture iterative e selettive OUTLINE Struttura selettiva Esempi Struttura
Fondamenti di Informatica AlgoBuild: Strutture selettive, iterative ed array Prof. Arcangelo Castiglione A.A. 2016/17 AlgoBuild : Strutture iterative e selettive OUTLINE Struttura selettiva Esempi Struttura
Tempo e spazio di calcolo (continua)
") Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Allineamento multiplo di sequenze
 Allineamento multiplo di sequenze Nicola Vitacolonna vitacolo@dimi.uniud.it http://www.dimi.uniud.it/~vitacolo Università degli Studi di Udine 16 aprile 2002 Two homologous sequences whisper... a full
Allineamento multiplo di sequenze Nicola Vitacolonna vitacolo@dimi.uniud.it http://www.dimi.uniud.it/~vitacolo Università degli Studi di Udine 16 aprile 2002 Two homologous sequences whisper... a full
Edit distance. v intner RIMDMDMMI wri t ers
 L'allineamento Edit distance Le operazioni permesse sono: I: insert (inserimento, inserzione) D: delete (cancellazione, delezione, rimozione) R: replacement (substition, sostituzione) M: match (corrispondenza,
L'allineamento Edit distance Le operazioni permesse sono: I: insert (inserimento, inserzione) D: delete (cancellazione, delezione, rimozione) R: replacement (substition, sostituzione) M: match (corrispondenza,
Lezione 12. Origine degli introni
 Lezione 12 Origine degli introni Nature Reviews Genetics 2006 Introni di gruppo I batteri, organelli identificati circa 1500 Introni di gruppo II identificati circa 200 Introni con spliceosomi genoma nucleare
Lezione 12 Origine degli introni Nature Reviews Genetics 2006 Introni di gruppo I batteri, organelli identificati circa 1500 Introni di gruppo II identificati circa 200 Introni con spliceosomi genoma nucleare
Corso di Perfezionamento
 Programmazione Dinamica 1 1 Dipartimento di Matematica e Informatica Università di Camerino 15 febbraio 2009 Tecniche di Programmazione Tecniche di progettazione di algoritmi: 1 Divide et Impera 2 Programmazione
Programmazione Dinamica 1 1 Dipartimento di Matematica e Informatica Università di Camerino 15 febbraio 2009 Tecniche di Programmazione Tecniche di progettazione di algoritmi: 1 Divide et Impera 2 Programmazione
07/01/2015. Come si ferma una macchina in corsa? Il terminatore. Terminazione intrinseca (rho-indipendente)
") Come si ferma una macchina in corsa? Il terminatore Terminazione intrinseca (rho-indipendente) Terminazione dipendente dal fattore Rho (r) 1 Operoni: gruppi di geni parte di una unica unità trascrizionale
Come si ferma una macchina in corsa? Il terminatore Terminazione intrinseca (rho-indipendente) Terminazione dipendente dal fattore Rho (r) 1 Operoni: gruppi di geni parte di una unica unità trascrizionale
Tempo e spazio di calcolo (continua)
") Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
Tempo e spazio di calcolo (continua) I numeri di Fibonacci come case study (applichiamo ad un esempio completo le tecniche illustrate nei lucidi precedenti) Abbiamo introdotto tecniche per la correttezza
RENDITE. Ricerca del tasso di una rendita
 RENDITE Ricerca del tasso di una rendita Un problema che si presenta spesso nelle applicazioni è quello di calcolare il tasso di interesse associato a una rendita quando siano note le altre grandezze 1
RENDITE Ricerca del tasso di una rendita Un problema che si presenta spesso nelle applicazioni è quello di calcolare il tasso di interesse associato a una rendita quando siano note le altre grandezze 1
DNA E PROTEINE IL DNA E RACCHIUSO NEL NUCLEO, MENTRE LA SINTESI PROTEICA SI SVOLGE NEL CITOPLASMA: COME VIENE TRASPORTATA L INFORMAZIONE?
 DNA E PROTEINE NUMEROSI DATI SUGGERISCONO CHE IL DNA SVOLGA IL SUO RUOLO GENETICO CONTROLLANDO LA SINTESI DELLE PROTEINE, IN PARTICOLARE DETERMINANDONE LA SEQUENZA IN AMINOACIDI E NECESSARIO RISPONDERE
DNA E PROTEINE NUMEROSI DATI SUGGERISCONO CHE IL DNA SVOLGA IL SUO RUOLO GENETICO CONTROLLANDO LA SINTESI DELLE PROTEINE, IN PARTICOLARE DETERMINANDONE LA SEQUENZA IN AMINOACIDI E NECESSARIO RISPONDERE
Programmazione dinamica
 Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Programmazione dinamica Fornisce l allineamento ottimale tra due sequenze semplici variazioni dell algoritmo producono allineamenti globali o locali l allineamento calcolato dipende dalla scelta di alcuni
Metodi per la riduzione della dimensionalità. Strumenti quantitativi per la gestione
 Metodi per la riduzione della dimensionalità Strumenti quantitativi per la gestione Emanuele Taufer file:///c:/users/emanuele.taufer/dropbox/3%20sqg/classes/6c_pca.html#(1) 1/25 Introduzione Gli approcci
Metodi per la riduzione della dimensionalità Strumenti quantitativi per la gestione Emanuele Taufer file:///c:/users/emanuele.taufer/dropbox/3%20sqg/classes/6c_pca.html#(1) 1/25 Introduzione Gli approcci
Algoritmi e Strutture Dati
 Algoritmi e Strutture Dati Capitolo 1 Un introduzione informale agli algoritmi Camil Demetrescu, Irene Finocchi, Giuseppe F. Italiano Definizione informale di algoritmo Insieme di istruzioni, definite
Algoritmi e Strutture Dati Capitolo 1 Un introduzione informale agli algoritmi Camil Demetrescu, Irene Finocchi, Giuseppe F. Italiano Definizione informale di algoritmo Insieme di istruzioni, definite
7 Disegni sperimentali ad un solo fattore. Giulio Vidotto Raffaele Cioffi
 7 Disegni sperimentali ad un solo fattore Giulio Vidotto Raffaele Cioffi Indice: 7.1 Veri esperimenti 7.2 Fattori livelli condizioni e trattamenti 7.3 Alcuni disegni sperimentali da evitare 7.4 Elementi
7 Disegni sperimentali ad un solo fattore Giulio Vidotto Raffaele Cioffi Indice: 7.1 Veri esperimenti 7.2 Fattori livelli condizioni e trattamenti 7.3 Alcuni disegni sperimentali da evitare 7.4 Elementi
Cluster Analysis. La Cluster Analysis è il processo attraverso il quale vengono individuati raggruppamenti dei dati. per modellare!
 La Cluster Analysis è il processo attraverso il quale vengono individuati raggruppamenti dei dati. Le tecniche di cluster analysis vengono usate per esplorare i dati e non per modellare! La cluster analysis
La Cluster Analysis è il processo attraverso il quale vengono individuati raggruppamenti dei dati. Le tecniche di cluster analysis vengono usate per esplorare i dati e non per modellare! La cluster analysis
Guida pratica per la creazione di un documento Word accessibile Sommario
 Guida pratica per la creazione di un documento Word accessibile Sommario Introduzione... 2 Proprietà... 2 Stili e formattazione... 2 Creazione di un sommario... 3 Collegamenti ipertestuali... 3 Colori...
Guida pratica per la creazione di un documento Word accessibile Sommario Introduzione... 2 Proprietà... 2 Stili e formattazione... 2 Creazione di un sommario... 3 Collegamenti ipertestuali... 3 Colori...
LE BASI DI DATI. Seconda parte La progettazione di database Relazionali SCHEMA LOGICO - Ristrutturazione dello schema concettuale
 LE BASI DI DATI Seconda parte La progettazione di database Relazionali SCHEMA LOGICO - Ristrutturazione dello schema concettuale LA PROGETTAZIONE LOGICA L'obiettivo della progettazione logica è quello
LE BASI DI DATI Seconda parte La progettazione di database Relazionali SCHEMA LOGICO - Ristrutturazione dello schema concettuale LA PROGETTAZIONE LOGICA L'obiettivo della progettazione logica è quello
Linguaggi, Traduttori e le Basi della Programmazione
 Corso di Laurea in Ingegneria Civile Politecnico di Bari Sede di Foggia Fondamenti di Informatica Anno Accademico 2011/2012 docente: Prof. Ing. Michele Salvemini Sommario Il Linguaggio I Linguaggi di Linguaggi
Corso di Laurea in Ingegneria Civile Politecnico di Bari Sede di Foggia Fondamenti di Informatica Anno Accademico 2011/2012 docente: Prof. Ing. Michele Salvemini Sommario Il Linguaggio I Linguaggi di Linguaggi
Rappresentazione grafica di sistemi atomici e molecolari: editor e visualizzatori ESERCITAZIONE 5
 Rappresentazione grafica di sistemi atomici e molecolari: editor e visualizzatori ESERCITAZIONE 5 Editor e visualizzatori Spesso è utile rappresentare graficamente la struttura atomica o molecolare del
Rappresentazione grafica di sistemi atomici e molecolari: editor e visualizzatori ESERCITAZIONE 5 Editor e visualizzatori Spesso è utile rappresentare graficamente la struttura atomica o molecolare del
Introduzione all analisi di arrays: clustering.
 Statistica per la Ricerca Sperimentale Introduzione all analisi di arrays: clustering. Lezione 2-14 Marzo 2006 Stefano Moretti Dipartimento di Matematica, Università di Genova e Unità di Epidemiologia
Statistica per la Ricerca Sperimentale Introduzione all analisi di arrays: clustering. Lezione 2-14 Marzo 2006 Stefano Moretti Dipartimento di Matematica, Università di Genova e Unità di Epidemiologia
Sommario. A proposito di A colpo d occhio 1. Novità di Access Primi passi con Access
 Sommario 1 2 3 A proposito di A colpo d occhio 1 Niente computerese!... 1 Una veloce panoramica... 2 Alcune osservazioni... 4 Per concludere... 4 Novità di Access 2010 5 Gestire le impostazioni e i file
Sommario 1 2 3 A proposito di A colpo d occhio 1 Niente computerese!... 1 Una veloce panoramica... 2 Alcune osservazioni... 4 Per concludere... 4 Novità di Access 2010 5 Gestire le impostazioni e i file
Metodo di Quine- McCluskey
 Metodo di Quine- McCluskey Maurizio Palesi Maurizio Palesi 1 Definizioni Date due funzioni f(x 1,x 2,,x n ) e g(x 1,x 2,,x n ) si dice che f copre g (oppure g implica f) e si scrive f g se f(x 1,x 2,,x
Metodo di Quine- McCluskey Maurizio Palesi Maurizio Palesi 1 Definizioni Date due funzioni f(x 1,x 2,,x n ) e g(x 1,x 2,,x n ) si dice che f copre g (oppure g implica f) e si scrive f g se f(x 1,x 2,,x
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre II. 4 Evoluzione e filogenesi
 Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre II 4 Evoluzione e filogenesi FILOGENETICA CS Definzione Studio delle relazioni evolutive tra
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre II 4 Evoluzione e filogenesi FILOGENETICA CS Definzione Studio delle relazioni evolutive tra
Riconoscimento e recupero dell informazione per bioinformatica
 Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
Riconoscimento e recupero dell informazione per bioinformatica Clustering: introduzione Manuele Bicego Corso di Laurea in Bioinformatica Dipartimento di Informatica - Università di Verona Una definizione
FUNZIONI BOOLEANE. Vero Falso
 FUNZIONI BOOLEANE Le funzioni booleane prendono il nome da Boole, un matematico che introdusse un formalismo che opera su variabili (dette variabili booleane o variabili logiche o asserzioni) che possono
FUNZIONI BOOLEANE Le funzioni booleane prendono il nome da Boole, un matematico che introdusse un formalismo che opera su variabili (dette variabili booleane o variabili logiche o asserzioni) che possono
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni:
: Allineamento di sequenze (parte 1) Antonella Meloni:") Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Lezione 2 (10/03/2010): Allineamento di sequenze (parte 1) Antonella Meloni: antonella.meloni@ifc.cnr.it Sequenza A= stringa formata da N simboli, dove i simboli apparterranno ad un certo alfabeto. A
Lezione 1. Le molecole di base che costituiscono la vita
 Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
Lezione 1 Le molecole di base che costituiscono la vita Le molecole dell ereditarietà 5 3 L informazione ereditaria di tutti gli organismi viventi, con l eccezione di alcuni virus, è a carico della molecola
Problemi e algoritmi. Il che cosa ed il come. Il che cosa ed il come. Il che cosa e il come
 Problemi e algoritmi Il che cosa e il come Problema: descrive che cosa si deve calcolare Specifica (di un algoritmo): descrive che cosa calcola un algoritmo Algoritmo: descrive come effettuare un calcolo
Problemi e algoritmi Il che cosa e il come Problema: descrive che cosa si deve calcolare Specifica (di un algoritmo): descrive che cosa calcola un algoritmo Algoritmo: descrive come effettuare un calcolo
Luigi Piroddi
 Automazione industriale dispense del corso 13. Reti di Petri: analisi strutturale sifoni e trappole Luigi Piroddi piroddi@elet.polimi.it Introduzione Abbiamo visto in precedenza il ruolo dei P-invarianti
Automazione industriale dispense del corso 13. Reti di Petri: analisi strutturale sifoni e trappole Luigi Piroddi piroddi@elet.polimi.it Introduzione Abbiamo visto in precedenza il ruolo dei P-invarianti
3. Confronto tra due sequenze
 3. Confronto tra due sequenze Esercizio 1: uso di DotLet Il programma DotLet è accessibile dal sito http://myhits.isb-sib.ch/cgi-bin/dotlet, dove può essere utilizzato attraverso un interfaccia utente
3. Confronto tra due sequenze Esercizio 1: uso di DotLet Il programma DotLet è accessibile dal sito http://myhits.isb-sib.ch/cgi-bin/dotlet, dove può essere utilizzato attraverso un interfaccia utente
Laboratorio di Bioinformatica I. Parte 2. Dott. Sergio Marin Vargas (2014 / 2015)
") Laboratorio di Bioinformatica I Banche dati Parte 2 Dott. Sergio Marin Vargas (2014 / 2015) Google Scholar https://scholar.google.it/ E un motore di ricerca di Google, specializzato nella ricerca di articoli
Laboratorio di Bioinformatica I Banche dati Parte 2 Dott. Sergio Marin Vargas (2014 / 2015) Google Scholar https://scholar.google.it/ E un motore di ricerca di Google, specializzato nella ricerca di articoli
Come si sceglie l algoritmo di allineamento? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione?
 Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Definire una chiave primaria. Microsoft Access. Definire una chiave primaria. Definire una chiave primaria. Definire una chiave primaria
 Microsoft Access Chiavi, struttura delle tabelle 1. Portare la tabella in Visualizzazione struttura Selezionare la tabella sulla quale si desidera intervenire nella finestra del database Poi: Fare clic
Microsoft Access Chiavi, struttura delle tabelle 1. Portare la tabella in Visualizzazione struttura Selezionare la tabella sulla quale si desidera intervenire nella finestra del database Poi: Fare clic
Ulteriori conoscenze di informatica Elementi di statistica Esercitazione3
 Ulteriori conoscenze di informatica Elementi di statistica Esercitazione3 Sui PC a disposizione sono istallati diversi sistemi operativi. All accensione scegliere Windows. Immettere Nome utente b## (##
Ulteriori conoscenze di informatica Elementi di statistica Esercitazione3 Sui PC a disposizione sono istallati diversi sistemi operativi. All accensione scegliere Windows. Immettere Nome utente b## (##
CURRICOLO DI MATEMATICA SCUOLA SECONDARIA DI PRIMO GRADO
 CURRICOLO DI MATEMATICA SCUOLA SECONDARIA DI PRIMO GRADO Nuclei tematici Il numero Traguardi per lo sviluppo della competenza - Muoversi con sicurezza nel calcolo anche con i numeri razionali e stimare
CURRICOLO DI MATEMATICA SCUOLA SECONDARIA DI PRIMO GRADO Nuclei tematici Il numero Traguardi per lo sviluppo della competenza - Muoversi con sicurezza nel calcolo anche con i numeri razionali e stimare
Evoluzione delle molecole biologiche
 Evoluzione delle molecole biologiche Un video (in inglese): clic Evoluzione delle emoglobine (I) Un esempio classico di evoluzione delle macromolecole biologiche è dato dall emoglobina(hb), la molecola
Evoluzione delle molecole biologiche Un video (in inglese): clic Evoluzione delle emoglobine (I) Un esempio classico di evoluzione delle macromolecole biologiche è dato dall emoglobina(hb), la molecola
Predizione di Struttura Secondaria di Proteine
 Laboratorio di Bioinformatica I Predizione di Struttura Secondaria di Proteine Dott. Sergio Marin Vargas (2014 / 2015) Struttura delle proteine Struttura Secondaria Elementi non strutturati (Non hanno
Laboratorio di Bioinformatica I Predizione di Struttura Secondaria di Proteine Dott. Sergio Marin Vargas (2014 / 2015) Struttura delle proteine Struttura Secondaria Elementi non strutturati (Non hanno
Database Modulo 6 CREAZIONE DI MASCHERE
 Database Modulo 6 CREAZIONE DI MASCHERE!1 Per la gestione dei dati strutturati è possibile utilizzare diverse modalità di visualizzazione. Si è analizzata sinora una rappresentazione di tabella (foglio
Database Modulo 6 CREAZIONE DI MASCHERE!1 Per la gestione dei dati strutturati è possibile utilizzare diverse modalità di visualizzazione. Si è analizzata sinora una rappresentazione di tabella (foglio
LE BASI DI DATI. Seconda parte La progettazione di database Relazionali SCHEMA LOGICO e SCHEMA FISICO Costruzione delle tabelle
 LE BASI DI DATI Seconda parte La progettazione di database Relazionali SCHEMA LOGICO e SCHEMA FISICO Costruzione delle tabelle LA PROGETTAZIONE LOGICA Lo scopo della fase di progettazione logica è quello
LE BASI DI DATI Seconda parte La progettazione di database Relazionali SCHEMA LOGICO e SCHEMA FISICO Costruzione delle tabelle LA PROGETTAZIONE LOGICA Lo scopo della fase di progettazione logica è quello
Corso di Ingegneria del Software. Modelli di produzione del software
 Corso di Ingegneria del Software a.a. 2009/2010 Mario Vacca mario.vacca1@istruzione.it 1. Concetti di base Sommario 2. 2.1 Modello a cascata 2.2 Modelli incrementali 2.3 2.4 Comparazione dei modelli 2.5
Corso di Ingegneria del Software a.a. 2009/2010 Mario Vacca mario.vacca1@istruzione.it 1. Concetti di base Sommario 2. 2.1 Modello a cascata 2.2 Modelli incrementali 2.3 2.4 Comparazione dei modelli 2.5
FILE E INDICI Architettura DBMS
 FILE E INDICI Architettura DBMS Giorgio Giacinto 2010 Database 2 Dati su dispositivi di memorizzazione esterni! Dischi! si può leggere qualunque pagina a costo medio fisso! Nastri! si possono leggere le
FILE E INDICI Architettura DBMS Giorgio Giacinto 2010 Database 2 Dati su dispositivi di memorizzazione esterni! Dischi! si può leggere qualunque pagina a costo medio fisso! Nastri! si possono leggere le
ISTRUZIONI UTILIZZO FILE EXCEL PER LA VALUTAZIONE DEL RSICHIO CHIMIMCO CON ALGORITMO MOVARISCH 2017
 Pag. 1 di 7 Sommario INTRODUZIONE... 2 USO DEL FOGLIO EXCEL... 2 CONCLUSIONI E DICHIARAZIONE DI ESCLUSIONE DI RESPONSABILITÀ... 7 Pag. 2 di 7 INTRODUZIONE Questo foglio Excel, contiene le istruzioni per
Pag. 1 di 7 Sommario INTRODUZIONE... 2 USO DEL FOGLIO EXCEL... 2 CONCLUSIONI E DICHIARAZIONE DI ESCLUSIONE DI RESPONSABILITÀ... 7 Pag. 2 di 7 INTRODUZIONE Questo foglio Excel, contiene le istruzioni per
Esercitazione n 2. Costruzione di grafici
 Esercitazione n 2 Costruzione di grafici I grafici I grafici sono rappresentazione di dati numerici e/o di funzioni. Devono facilitare all utente la visualizzazione e la comprensione dei numeri e del fenomeno
Esercitazione n 2 Costruzione di grafici I grafici I grafici sono rappresentazione di dati numerici e/o di funzioni. Devono facilitare all utente la visualizzazione e la comprensione dei numeri e del fenomeno
Lezione 6. Analisi di sequenze biologiche e ricerche in database
 Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Gasparini Alessandra BIM
 Gasparini Alessandra 592026-BIM Pfam è una banca dati di famiglie proteiche creata dal Wellcome Trust Sanger Institute a partire dal database Pfamseq basato su Uniprot. Le famiglie proteiche di Pfam sono
Gasparini Alessandra 592026-BIM Pfam è una banca dati di famiglie proteiche creata dal Wellcome Trust Sanger Institute a partire dal database Pfamseq basato su Uniprot. Le famiglie proteiche di Pfam sono
RELAZIONE di BIOLOGIA MOLECOLARE
 NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
NOME: Marini Selena MATRICOLA: 592330 RELAZIONE di BIOLOGIA MOLECOLARE CHE ORGANISMO MODELLO È DICTYOSTELIUM? CHE RISORSE BIOINFORMATICHE AGEVOLANO I RICERCATORI CHE LO STUDIANO? Dictyostelium è un genere
Metodo di Quine e MC-Cluskey 2/2 Prof. Mario Cannataro Università degli Studi Magna Graecia di Catanzaro
 Fondamenti di Informatica II Ingegneria Informatica e Biomedica I anno, II semestre A.A. 2005/2006 Metodo di Quine e MC-Cluskey 2/2 Prof. Mario Cannataro Università degli Studi Magna Graecia di Catanzaro
Fondamenti di Informatica II Ingegneria Informatica e Biomedica I anno, II semestre A.A. 2005/2006 Metodo di Quine e MC-Cluskey 2/2 Prof. Mario Cannataro Università degli Studi Magna Graecia di Catanzaro
I giochi con avversario. I giochi con avversario. Introduzione. Giochi come problemi di ricerca. Il gioco del NIM.
 I giochi con avversario I giochi con avversario Maria Simi a.a. 26/27 Regole semplici e formalizzabili eterministici, due giocatori, turni alterni, zero-sum, informazione perfetta (ambiente accessibile)
I giochi con avversario I giochi con avversario Maria Simi a.a. 26/27 Regole semplici e formalizzabili eterministici, due giocatori, turni alterni, zero-sum, informazione perfetta (ambiente accessibile)
La struttura covalente delle proteine (la sequenza amminoacidica)
") La struttura covalente delle proteine (la sequenza amminoacidica) Sequenza amminoacidica dell ormone insulina bovino (Frederick Sanger, 1953) Il primo passo per determinare la sequenza di un peptide è
La struttura covalente delle proteine (la sequenza amminoacidica) Sequenza amminoacidica dell ormone insulina bovino (Frederick Sanger, 1953) Il primo passo per determinare la sequenza di un peptide è
1 Schemi alle differenze finite per funzioni di una variabile
 Introduzione In questa dispensa vengono forniti alcuni elementi di base per la soluzione di equazioni alle derivate parziali che governano problemi al contorno. A questo scopo si introducono, in forma
Introduzione In questa dispensa vengono forniti alcuni elementi di base per la soluzione di equazioni alle derivate parziali che governano problemi al contorno. A questo scopo si introducono, in forma
Tesina di Biologia Molecolare II
 MELATO GIULIA 595033 Tesina di Biologia Molecolare II Mostra un albero filogenetico con la relazione tra Uomo, Topo e Ratto. Che banca dati è disponibile per quest'ultimo organismo? Descrivi alcune caratteristiche
MELATO GIULIA 595033 Tesina di Biologia Molecolare II Mostra un albero filogenetico con la relazione tra Uomo, Topo e Ratto. Che banca dati è disponibile per quest'ultimo organismo? Descrivi alcune caratteristiche