Protein folding. Un gran numero di interazioni deboli + ΔH
|
|
|
- Gloria Basso
- 6 anni fa
- Visualizzazioni
Transcript
1



2 Protein folding -ΔS Un gran numero di interazioni deboli +ΔS + ΔH E r
3 Protein structure modelling: A digression I polimeri (inclusi quelli di amino acidi) in generale non hanno una struttura unica. Le proteine sono polimeri speciali perché sono stati selezionati dall evoluzione per svolgere una determinata funzione. La loro stabilità è solo marginale. La probabilità che una sequenza casuale di amino acidi soddisfi queste condizioni è molto bassa. This is a rare event: in fact, it is a miracle*! *Aliosha Finkelstein
4 Immaginiamo... Mutazione di un amino acido Protein structure modelling: A digression
5 Immaginiamo... Mutazione di un amino acido Protein structure modelling: A digression La proteina non si struttura più Il risultato più probabile
6 Immaginiamo... Mutazione di un amino acido Protein structure modelling: A digression La proteina non si struttura più La nuova sequenza assume una struttura completamente diversa, stabile e funzionale Il risultato più probabile Un miracolo 2

7 Immaginiamo... Mutazione di un amino acido Protein structure modelling: A digression La proteina non si struttura più Il risultato più probabile La mutazione si accomoda con distorsioni locali Il solo risultato possibile se c'è pressione evolutiva La nuova sequenza assume una struttura completamente diversa, stabile e funzionale Un miracolo 2
8 In conclusione La fisica delle proteine e l evoluzione garantiscono che proteine omologhe hanno struttura simile
9 In conclusione La fisica delle proteine e l evoluzione garantiscono che proteine omologhe hanno struttura simile
10 Quanto simili? Misurare la similarità di struttura Possiamo misurare la distanza media tra atomi corrispondenti
11 Root mean square deviation (RMSD) RMSD = 1 N N [(x i x ' i ) 2 + (y i y ' i ) 2 + (z i z ' i ) 2 ] i=1

12 L RMSD puo non essere la misura ideale
13 GDT-TS GDT TS =100 n 1 + n 2 + n 3 + n 4 4L n i = numero di aminoacidi allineati distanti meno di i Å L = numero di aminoacidi allineati
14 TM-score
15 Sovrapposizione Dati due insiemi di punti A=(a 1, a 2,... a n ) e B= (b 1, b 2,..., b m ) occorre trovare la trasformazione G che minimizza la distanza: min G Con P=Q, d di solito è rmsd { d[a(p) G[B(Q)] }
16 Quindi Occorre trovare la corrispondenza tra i punti di A e i punti di B (NP-hard) Trovare la trasformazione (rotazione e traslazione) (O(n))
17 Sovrapposizione strutturale Sovrapporre e misurare la distanza tra due strutture con la stessa sequenza è semplice, basta minimizzare la distanza fra atomi corrispondenti Negli altri casi: Si può usare un allineamento di sequenza per ottenere la corrispondenza Si può cercare la migliore corrispondenza e la trasformazione contemporaneamente
18 Atomi corrispondenti La catena polipeptidica è formata da una parte ripetitiva (catena principale o backbone) e da una parte variabile (catene laterali o residui). Se le sequenze delle proteine sono diverse, dobbiamo limitarci al backbone.
19 Metodi DALI: Rappresenta le strutture come matrici di distanza tra gli atomi Ca Holm and Sander. Protein structure comparison by alignment of distance matrices. J Mol Biol 1993, 233: CE (Combinatorial extension): Usa caratteristiche di geometria locale e poi le unisce in un percorso ottimale Shindyalov and Bourne, Protein structure alignment by incremental combinatorial extension (CE) of optimal path. Prot Eng, 1998, 11: SSAP (Sequential Structure Alignment Program ): Usa un algortimo di doppia programmazione dinamica Taylor WR, Orengo CA. 1989b. Protein structure alignment. J Mol Biol 208:l-22 VAST (Vector Alignment Search Tool ), TM-align etc.


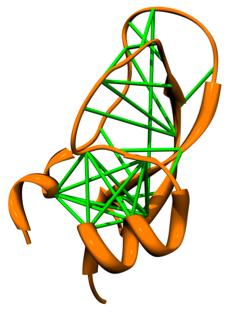
20 Mappa di contatti
21 DALI Divide la struttura in frammenti di 6 aminoacidi e costruisce una matrice di contatto per ogni frammento Confronta sottomatrici 6x6 Ottimizza il punteggio finale di sovrapposizione con MC
22 DALI: Sottomatrici Image from Mark Maciejewski at UConn
 le possibili")

23 Confronta (tutte?) le possibili combinazioni di matrici A d ij B d ij d ij A d ij B
24 Problemino 100 amino acidi -> 96 esapeptidi = 95 2 /2 = matrici 150 amino acidi -> 145 esapeptidi = /2 = matrici confronti
25 In realtà Confronta la matrice di una proteina con tutte quelle dell altra Ripete per tutte le matrici Ordina le coppie per similarità Unisce matrici consecutive e conserva solo la matrice del set con maggiore similarità




26 DALI In questo modo conserva solo una matrice per ogni set
27 DALI Ci sono ancora troppe coppie Confronta le coppie a caso Mantiene solo le coppie con un punteggio positivo Finisci quando ne ha 80,000 Prende le 40,000 migliori Usa Monte Carlo per ottimizzare la sovrapposizione finale Per valutare la significatività usa lo Z-score
28 Quanto simili sono le proteine omologhe RMSD Percentuale di identità di sequenza Dipende dalla distanza evolutiva (che possiamo stimare dalla percentuale di identità di sequenza) Chothia and Lesk, EMBO J., 1986
 come prima approssimazione della")

29 Protein structure modelling: Possiamo usare la struttura A digression della proteina omologa (templato) come prima approssimazione della struttura della nostra proteina (target) Protein No. FLAV_CLOBE 1 A... I V Y W S G T G N T E K M A E CYSJ_THIRO 2 A. I T I L F G S Q T G N A K A V A E

30 Date due strutture proteiche sovrapposte, si può dedurre la corrispondenza tra i loro amino acidi AVSERT ALSDRS
31 Data la corrispondenza tra gli amino acidi di sue proteine (allineamento) si può dedurre la corrispondenza tra i loro atomi in tre dimensioni AVSERT ALSDRS

32
33 T GFFS VSD? ATA VTE LSA KV G V V S D E V V A A S L A T A
34 GFFS VSD? ATA VTE LSA KV
35 GFFS VSD? ATA VTE LSA KV

36 GFFS VSD? ATA VTE LSA KV

37 GFFS VSD? ATA VTE LSA KV
38 L allineamento corretto è essenziale Protein No. FLAV_CLOBE 1 A... I V Y W S G T G N T E K M A E CYSJ_THIRO 2 A. I T I L F G S Q T G N A K A V A E CYSJ_ECOLI 3 A. I T I I S A S Q T G N A R R V A E NOS2_CHICK 4 A K V T V I Y A T E T G K S E T L A N NOS2_ONCMY 5 A.. T V L Y A T E T G K S Q T L A Q NOS1_RABIT 6 A K A T I L Y A T E T G K S Q A Y A K NOS3_HUMAN 7 A K A T I L Y G S E T G R A Q S Y A Q NOS_RHOPR 8 A K A T I L F A T E T G K S E M Y A R NOS_ANOST 9 A K A T V L Y A T E T G R S E Q Y A R NOS_LYMST 10 A K C S I F Y A T E T G R S E R F A R NCPR_HUMAN 11 A N I I V F Y G S Q T G T A E E F A N NCPR_CANTR 12 A N T L L L F G S Q T G T A E D Y A N NCPR_SCHPO 13 A. A A V F F G S Q T G T A E D F A Y NCPR_YEAST 14 A N Y L V L Y A S Q T G T A E D Y A K FLAV_DESSA 15 A K S L I V Y G S T T G N T E T A A E FLAV_DESGI 16 A K A L I V Y G S T T G N T E G V A E
39 Occorre modellare le regioni strutturalmente divergenti Per regioni piccole si possono usare regole basate sulle sequenze Si possono esplorare le possibili conformazioni e valutarne l energia nel contesto della proteina Si possono utilizzare metodi euristici
40 Un metodo euristico Numero di amino acidi d
41 Un metodo euristico Numero di amino acidi d
42 Un metodo euristico Numero di amino acidi d
43 Un metodo euristico Numero di amino acidi d

44 Modello del backbone
45 Le catene laterali

46 Le catene laterali

47 Le catene laterali possono assumere conformazioni diverse Esplorarle tutte è tecnicamente impossibile
48 Non tutte le possibili conformazioni sono equiprobabili 15% 22% 25% 33% Possiamo stimare le probabilità analizzando la frequenza nelle strutture note


49 Librerie di rotameri 15% 22% 33% 25%


50 Librerie di rotameri Esploriamo solo le conformazioni presenti nella libreria seguendo l ordine della loro probabilità Possiamo usare il dead end elimination algorithm identificando le conformazioni che non possono far parte del minimo globale.
51 Altra possibilità Spesso abbiamo più di una proteina di struttura nota omologa alla nostra (templati) Ciascun templato ha una diversa distanza evolutiva dalla nostra proteina target e quindi sarà più o meno simile in struttura Templato 1 Templato 2
52 Altra possibilità Spesso abbiamo più di una proteina di struttura nota omologa alla nostra (templati) Ciascun templato ha una diversa distanza evolutiva dalla nostra proteina target e quindi sarà più o meno simile in struttura Templato 1 Templato 2 Modello
53 Altra possibilità Spesso abbiamo più di una proteina di struttura nota omologa alla nostra (templati) Ciascun templato ha una diversa distanza evolutiva dalla nostra proteina target e quindi sarà più o meno simile in struttura Possiamo ottenere le distribuzioni di probabilità (PDF) di osservare certe differenze tra distanze, angoli, etc, in funzione della distanza evolutiva

54 Esempio: distanze tra i Ca g: Distanza media da una gap i: percentuale di identità a: accessibilità al solvente d : distanza

55 Modeller Allinea sequenze e strutture Estrai i vincoli spaziali Ottimizza soddisfacendo i vincoli
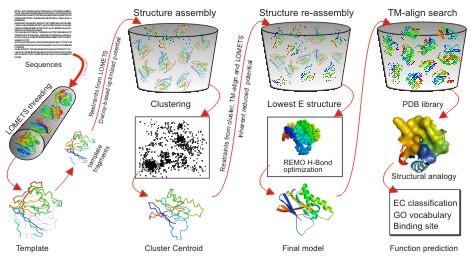
56 Tasser
57 Se non abbiamo un templato Metodi ab initio non sono ancora sufficientemente accurati I metodi più accurati al momento sono basati su frammenti
58 Metodi basati su frammenti AVGIFRAAVCTRGVAKAVDFVP AVGIFR AAVCTR GVAKAVDF Dividiamo la sequenza in frammenti
59 Metodi basati su frammenti AVGIFRAAVCTRGVAKAVDFVP AVGIFR AAVCTR GVAKAVDF Dividiamo la sequenza in frammenti Per ciascun frammento cerchiamo LE regioni di proteine di struttura nota con similarità di sequenza
60 Metodi basati su frammenti AVGIFRAAVCTRGVAKAVDFVP AVGIFR AAVCTR GVAKAVDF Combiniamole casualmente
61 Metodi basati su frammenti AVGIFRAAVCTRGVAKAVDFVP AVGIFR AAVCTR GVAKAVDF Combiniamole casualmente
62 Metodi basati su frammenti AVGIFRAAVCTRGVAKAVDFVP AVGIFR AAVCTR GVAKAVDF Combiniamole casualmente
63 Metodi basati su frammenti OTTIMIZZAZIONE: Monte Carlo, Simulated annealing, algoritmi genetici,...
64 Potenziali di coppia
65 [ ] " " # $ % % & ' = atoms bonded non C ij j i C ij ij C ij ij C angles dihedrals eq C eq bonds C b C r q q r B r A n K K b b K E, 0 6, 12, 2 2 ) cos( 1 2 ) ( ) ( ε γ φ φ θ θ θ Energia
66 Metodi basati su frammenti Si distinguono per: La dimensione dei frammenti Il data base di frammenti La metodologia di selezione dei frammenti La funzione o le funzioni energia
 Templati strutturali Domini strutturali Identificazione di domini Modello finale Assemblaggio e")
67 Metodi basati su frammenti In alcuni casi usano domini o arrangiamenti frequenti di strutture secondarie (strutture supersecondarie) Templati strutturali Domini strutturali Identificazione di domini Modello finale Assemblaggio e ottimizzazione
68 Possibile schema Frammenti candidati Selezione dei frammenti Sequenza di amino acidi Assemblaggio dei frammenti Modelli Ottimizzazione Valutazione Lista ordinata
69 Possibile schema Frammenti candidati Selezione dei frammenti Sequenza di amino acidi Assemblaggio dei frammenti Predizioni Modelli Ottimizzazione Dati sperimentali Valutazione Lista ordinata
70 Predizioni Struttura secondaria Basati su metodi di apprendimento automatico Accessibilità al solvente Basati su metodi di apprendimento automatico Contatti...
Esempio: distanze tra i Cα. g: Distanza media da una gap i: percentuale di iden7tà a: accessibilità al solvente d : distanza
 Altra possibilità Spesso abbiamo più di una proteina di stru4ura nota omologa alla nostra (templa7) Ciascun templato ha una diversa distanza evolu7va dalla nostra proteina target e quindi sarà più o meno
Altra possibilità Spesso abbiamo più di una proteina di stru4ura nota omologa alla nostra (templa7) Ciascun templato ha una diversa distanza evolu7va dalla nostra proteina target e quindi sarà più o meno
Ricerca di omologhi. La sequenza di cui vogliamo trovare gli omologhi viene de6a query.
 Ricerca di omologhi La sequenza di cui vogliamo trovare gli omologhi viene de6a query. Dobbiamo cercare i suoi omologhi in una banca da= di sequenze (qualche decina di milioni) Allineamento con ciascuna
Ricerca di omologhi La sequenza di cui vogliamo trovare gli omologhi viene de6a query. Dobbiamo cercare i suoi omologhi in una banca da= di sequenze (qualche decina di milioni) Allineamento con ciascuna
Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Quarta lezione. 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST
 Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Quarta lezione 1. Ricerca di omologhe in banche dati. 2. Programmi per la ricerca: FASTA BLAST Ricerca di omologhe in banche dati Proteina vs. proteine Gene (traduzione in aa) vs. proteine Gene vs. geni
Predizione della struttura terziaria
 Predizione della struttura terziaria Metodi di predizione La predizione della struttura tridimensionale è di gran lunga la predizione più complessa che si possa fare su una proteina. Esistono 3 metodi
Predizione della struttura terziaria Metodi di predizione La predizione della struttura tridimensionale è di gran lunga la predizione più complessa che si possa fare su una proteina. Esistono 3 metodi
Predire la struttura terziaria
 Predire la struttura terziaria E di gran lunga la predizione più complessa che si possa fare su una proteina. Esistono 3 metodi principali di predizione: 1 - Homology modelling: se si conoscono proteine
Predire la struttura terziaria E di gran lunga la predizione più complessa che si possa fare su una proteina. Esistono 3 metodi principali di predizione: 1 - Homology modelling: se si conoscono proteine
E il server più utilizzato, permette di tracciare tutte le operazioni che svolge e di impostare alcuni parametri importanti per il risultato finale.
 Homology Modelling Homology modelling L omology modeling delle proteine è il tipo di predizione di struttura terziaria più semplice ed affidabile. Viene richiesta soltanto una (o più) sequenze di riferimento
Homology Modelling Homology modelling L omology modeling delle proteine è il tipo di predizione di struttura terziaria più semplice ed affidabile. Viene richiesta soltanto una (o più) sequenze di riferimento
q xi Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p i >0;
 Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Modelli probabilis-ci Lanciando un dado abbiamo sei parametri p1 p6 p i >0; 6! i=1 p i =1 Sequenza di dna/proteine x con probabilita q x Probabilita dell intera sequenza n " i!1 q xi Massima verosimiglianza
Proprietà comuni. Il gruppo α-carbossilico b è un acido più forte del gruppo carbossilico degli acidi alifatici
 Gli aminoacidi Proprietà comuni Il gruppo α-carbossilico b è un acido più forte del gruppo carbossilico degli acidi alifatici paragonabili Il gruppo α-aminico è un acido più forte (o una base più debole
Gli aminoacidi Proprietà comuni Il gruppo α-carbossilico b è un acido più forte del gruppo carbossilico degli acidi alifatici paragonabili Il gruppo α-aminico è un acido più forte (o una base più debole
Chimica Biologica A.A α-elica foglietto β reverse turn
 Chimica Biologica A.A. 2010-2011 α-elica foglietto β reverse turn Str. Secondaria sperimentalmente osservata: Si distinguono fondamentalmente tre tipi di strutture secondarie: α elica foglietto β reverse
Chimica Biologica A.A. 2010-2011 α-elica foglietto β reverse turn Str. Secondaria sperimentalmente osservata: Si distinguono fondamentalmente tre tipi di strutture secondarie: α elica foglietto β reverse
Ricerca di omologia di sequenza
 Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
Ricerca di omologia di sequenza RICERCA DI OMOLOGIA DI SEQUENZA := Data una sequenza (query), una banca dati, un sistema per il confronto e una soglia statistica trovare le sequenze della banca più somiglianti
CENNI SUL TIPO DI FORZE
 CENNI SUL TIPO DI FORZE Forze deboli che influenzano la struttura delle proteine: le interazioni di van der Waals repulsione attrazione Forze attrattive dovute a interazioni istantanee che si generano
CENNI SUL TIPO DI FORZE Forze deboli che influenzano la struttura delle proteine: le interazioni di van der Waals repulsione attrazione Forze attrattive dovute a interazioni istantanee che si generano
8. Sovrapposizione e confronto di strutture proteiche
 8. Sovrapposizione e confronto di strutture proteiche In questa esercitazione utilizzeremo il programma SwissPdbViewer (versione 4.0.1) per effettuare la sovrapposizione e il confronto di strutture proteiche.
8. Sovrapposizione e confronto di strutture proteiche In questa esercitazione utilizzeremo il programma SwissPdbViewer (versione 4.0.1) per effettuare la sovrapposizione e il confronto di strutture proteiche.
E il server più utilizzato, permette di tracciare tutte le operazioni che svolge e di impostare alcuni parametri importanti per il risultato finale.
 Homology modelling L omology modeling delle proteine è il tipo di predizione di struttura terziaria più semplice ed affidabile. Viene richiesta soltanto una (o più) sequenze di riferimento su cui modellare
Homology modelling L omology modeling delle proteine è il tipo di predizione di struttura terziaria più semplice ed affidabile. Viene richiesta soltanto una (o più) sequenze di riferimento su cui modellare
BLAST. W = word size T = threshold X = elongation S = HSP threshold
 BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
BLAST Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando l algoritmo di Altschul et al. Anche Blast, come FASTA, funziona: 1. scomponendo la sequenza query
FASTA: Lipman & Pearson (1985) BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento
 BLAST: Altshul (1990) Algoritmi EURISTICI di allineamento") Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Algoritmi EURISTICI di allineamento Sono nati insieme alle banche dati, con lo scopo di permettere una ricerca per similarità rapida anche se meno accurata contro le migliaia di sequenze depositate. Attualmente
Macromolecole Biologiche. La struttura secondaria (II)
") La struttura secondaria (II) Nello stesso anno (1951) in cui proposero l α elica, Pauling e Corey postularono anche l esistenza di un altra struttura secondaria: il foglietto β (β-sheet). Dopo l α elica,
La struttura secondaria (II) Nello stesso anno (1951) in cui proposero l α elica, Pauling e Corey postularono anche l esistenza di un altra struttura secondaria: il foglietto β (β-sheet). Dopo l α elica,
InfoBioLab I ENTREZ. ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche
 InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
InfoBioLab I ES 1: Ricerca di sequenze di aminoacidi in banche dati biologiche Esercizio 1 - obiettivi: Ricerca di 2 proteine in ENTREZ Salva i flat file che descrivono le 2 proteine in formato testo Importa
Si osserva il comportamento dei protoni La proteina è in soluzione
 Risonanza magnetica nucleare Si osserva il comportamento dei protoni La proteina è in soluzione Risonanza magnetica nucleare Si osserva il comportamento dei protoni La proteina è in soluzione Si assegnano
Risonanza magnetica nucleare Si osserva il comportamento dei protoni La proteina è in soluzione Risonanza magnetica nucleare Si osserva il comportamento dei protoni La proteina è in soluzione Si assegnano
Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI. (Visione 3D)
") ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI () Structure From Motion Date m immagini di n punti 3D (fissi) Stimare le m matrici di proiezione P i e gli n vettori X j date le mn corrispondenze x ij SFM
ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI () Structure From Motion Date m immagini di n punti 3D (fissi) Stimare le m matrici di proiezione P i e gli n vettori X j date le mn corrispondenze x ij SFM
Allineamenti di sequenze: concetti e algoritmi
 Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Allineamenti di sequenze: concetti e algoritmi 1 globine: a- b- mioglobina Precoce esempio di allineamento di sequenza: globine (1961) H.C. Watson and J.C. Kendrew, Comparison Between the Amino-Acid Sequences
Structural Genomics. Grand goal : asssegnare una struttura ad ogni sequenza esistente.
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Se la funzione è analiticamente invertibile, estratto q, si può ricavare x = x(q).
.") La tecnica Monte Carlo Il metodo Monte Carlo è basato sulla scelta di eventi fisici con una probabilità di accadimento nota a priori. sia p(x) la distribuzione di probabilità con la quale si manifesta
La tecnica Monte Carlo Il metodo Monte Carlo è basato sulla scelta di eventi fisici con una probabilità di accadimento nota a priori. sia p(x) la distribuzione di probabilità con la quale si manifesta
30/10/2015 LIVELLI DI ORGANIZZAZIONE STRUTTURALE DELLE PROTEINE
 LIVELLI DI ORGANIZZAZIONE STRUTTURALE DELLE PROTEINE 1 CARATTERISTICHE DEL LEGAME PEPTIDICO lunghezza intermedia tra un legame singolo e uno doppio ibrido di risonanza per il parziale carattere di doppio
LIVELLI DI ORGANIZZAZIONE STRUTTURALE DELLE PROTEINE 1 CARATTERISTICHE DEL LEGAME PEPTIDICO lunghezza intermedia tra un legame singolo e uno doppio ibrido di risonanza per il parziale carattere di doppio
Modelling. Perché considerare la struttura 3D di una proteina
 Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Modelling Perché considerare la struttura 3D di una proteina Implicazioni in vari campi : biologia, evoluzione, biotecnologie, medicina, chimica farmaceutica... Metodi di studio della struttura di una
Aminoacidi. Gli α-aminoacidi sono molecole con almeno due gruppi funzionali legati al carbonio α
 Aminoacidi Gli α-aminoacidi sono molecole con almeno due gruppi funzionali legati al carbonio α 1 Isomeria ottica Tutti gli AA, esclusa la glicina, presentano almeno un atomo di carbonio asimmetrico, il
Aminoacidi Gli α-aminoacidi sono molecole con almeno due gruppi funzionali legati al carbonio α 1 Isomeria ottica Tutti gli AA, esclusa la glicina, presentano almeno un atomo di carbonio asimmetrico, il
Problemi e algoritmi. Il che cosa ed il come. Il che cosa ed il come. Il che cosa e il come
 Problemi e algoritmi Il che cosa e il come Problema: descrive che cosa si deve calcolare Specifica (di un algoritmo): descrive che cosa calcola un algoritmo Algoritmo: descrive come effettuare un calcolo
Problemi e algoritmi Il che cosa e il come Problema: descrive che cosa si deve calcolare Specifica (di un algoritmo): descrive che cosa calcola un algoritmo Algoritmo: descrive come effettuare un calcolo
Algoritmi di Allineamento
 Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Algoritmi di Allineamento CORSO DI BIOINFORMATICA Corso di Laurea in Biotecnologie Università Magna Graecia Catanzaro Outline Similarità Allineamento Omologia Allineamento di Coppie di Sequenze Allineamento
Il gruppo peptidico ha una struttura rigida e planare, dovuta al parziale. legame peptidico. O O - N N + H H
 Il legame peptidico Il gruppo peptidico ha una struttura rigida e planare, dovuta al parziale (~40 %) carattere di doppio legame del legame peptidico. O O - C C N N + H H Il legame peptidico pp Il legame
Il legame peptidico Il gruppo peptidico ha una struttura rigida e planare, dovuta al parziale (~40 %) carattere di doppio legame del legame peptidico. O O - C C N N + H H Il legame peptidico pp Il legame
Diagramma di Ramachandran
 Chimica Biologica A.A. 2010-2011 Diagramma di Ramachandran Diagramma di Ramachandran Catena polipeptidica La formazione in successione di legami peptidici genera la cosiddetta catena principale o scheletro
Chimica Biologica A.A. 2010-2011 Diagramma di Ramachandran Diagramma di Ramachandran Catena polipeptidica La formazione in successione di legami peptidici genera la cosiddetta catena principale o scheletro
Allineamenti a coppie
 Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Laboratorio di Bioinformatica I Allineamenti a coppie Dott. Sergio Marin Vargas (2014 / 2015) ExPASy Bioinformatics Resource Portal (SIB) http://www.expasy.org/ Il sito http://myhits.isb-sib.ch/cgi-bin/dotlet
Relazione sequenza-struttura e funzione
 Biotecnologie applicate alla progettazione e sviluppo di molecole biologicamente attive A.A. 2010-2011 Modulo di Biologia Strutturale Relazione sequenza-struttura e funzione Marco Nardini Dipartimento
Biotecnologie applicate alla progettazione e sviluppo di molecole biologicamente attive A.A. 2010-2011 Modulo di Biologia Strutturale Relazione sequenza-struttura e funzione Marco Nardini Dipartimento
A W T V A S A V R T S I A Y T V A A A V R T S I A Y T V A A A V L T S I
 COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
COME CALCOLARE IL PUNTEIO DI UN ALLINEAMENTO? Il problema del calcolo del punteggio di un allineamento può essere considerato in due modi diversi che, però, sono le due facce di una stessa medaglia al
FASTA. Lezione del
 FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
FASTA Lezione del 10.03.2016 Omologia vs Similarità Quando si confrontano due sequenze o strutture si usano spesso indifferentemente i termini somiglianza o omologia per indicare che esiste un rapporto
sono le unità monomeriche che costituiscono le proteine hanno tutti una struttura comune
 AMINO ACIDI sono le unità monomeriche che costituiscono le proteine sono 20 hanno tutti una struttura comune sono asimmetrici La carica di un amino acido dipende dal ph Classificazione amino acidi Glicina
AMINO ACIDI sono le unità monomeriche che costituiscono le proteine sono 20 hanno tutti una struttura comune sono asimmetrici La carica di un amino acido dipende dal ph Classificazione amino acidi Glicina
Markov Chains and Markov Chain Monte Carlo (MCMC)
") Markov Chains and Markov Chain Monte Carlo (MCMC) Alberto Garfagnini Università degli studi di Padova December 11, 2013 Catene di Markov Discrete dato un valore x t del sistema ad un istante di tempo fissato,
Markov Chains and Markov Chain Monte Carlo (MCMC) Alberto Garfagnini Università degli studi di Padova December 11, 2013 Catene di Markov Discrete dato un valore x t del sistema ad un istante di tempo fissato,
UNIVERSITÀ DEGLI STUDI DI MILANO. Bioinformatica. A.A semestre I. Allineamento veloce (euristiche)
") Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Docente: Matteo Re UNIVERSITÀ DEGLI STUDI DI MILANO C.d.l. Informatica Bioinformatica A.A. 2013-2014 semestre I 3 Allineamento veloce (euristiche) Banche dati primarie e secondarie Esistono due categorie
Macromolecole Biologiche. La struttura secondaria (III)
") La struttura secondaria (III) Reverse turn Le proteine globulari hanno una forma compatta, dovuta a numerose inversioni della direzione della catena polipeptidica che le compone. Molte di queste inversioni
La struttura secondaria (III) Reverse turn Le proteine globulari hanno una forma compatta, dovuta a numerose inversioni della direzione della catena polipeptidica che le compone. Molte di queste inversioni
Introduzione al Metodo del Simplesso. 1 Soluzioni di base e problemi in forma standard
 Introduzione al Metodo del Simplesso Giacomo Zambelli 1 Soluzioni di base e problemi in forma standard Consideriamo il seguente problema di programmazione lineare (PL), relativo all esempio di produzione
Introduzione al Metodo del Simplesso Giacomo Zambelli 1 Soluzioni di base e problemi in forma standard Consideriamo il seguente problema di programmazione lineare (PL), relativo all esempio di produzione
La ricerca di similarità in banche dati
 La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
La ricerca di similarità in banche dati Uno dei problemi più comunemente affrontati con metodi bioinformatici è quello di trovare omologie di sequenza interrogando una banca dati. L idea di base è che
gruppo amminico Gli aminoacidi polimerizzano durante la sintesi delle proteine mediante la formazione di legami peptidici. gruppo carbossilico
 gruppo amminico Gli aminoacidi polimerizzano durante la sintesi delle proteine mediante la formazione di legami peptidici. gruppo carbossilico Il legame peptidico si ha quando il gruppo carbossilico (-
gruppo amminico Gli aminoacidi polimerizzano durante la sintesi delle proteine mediante la formazione di legami peptidici. gruppo carbossilico Il legame peptidico si ha quando il gruppo carbossilico (-
Formazione del legame peptidico:
 Formazione del legame peptidico: Planare, ha una forza intermedia tra il legame semplice ed il legame doppio. 2^ lezione N R C C O O O + R N R C O C O O N R C C N C C O Ogni piano delle unità peptidiche
Formazione del legame peptidico: Planare, ha una forza intermedia tra il legame semplice ed il legame doppio. 2^ lezione N R C C O O O + R N R C O C O O N R C C N C C O Ogni piano delle unità peptidiche
La struttura delle proteine e e descritta da quattro livelli di organizzazione
 La struttura delle proteine e e descritta da quattro livelli di organizzazione Struttura Primaria- - la sequenza di aminoacidi Struttura Secondaria - strutture locali stabilizzate da legami H che coinvolgono
La struttura delle proteine e e descritta da quattro livelli di organizzazione Struttura Primaria- - la sequenza di aminoacidi Struttura Secondaria - strutture locali stabilizzate da legami H che coinvolgono
COMPORTAMENTO ANFOTERO DEGLI AA
 Proprietà acido-basiche degli aminoacidi FORMA NON IONICA Non esiste a nessun valore di ph FORMA ZWITTERIONICA È la forma prevalente a ph 7 COMPORTAMENTO ANFOTERO DEGLI AA CARICA NETTA +1 CARICA NETTA
Proprietà acido-basiche degli aminoacidi FORMA NON IONICA Non esiste a nessun valore di ph FORMA ZWITTERIONICA È la forma prevalente a ph 7 COMPORTAMENTO ANFOTERO DEGLI AA CARICA NETTA +1 CARICA NETTA
ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI. (Visione 3D)
") ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI () Calibrazione intrinseca Spesso risulta utile calibrare la sola componente intrinseca di un sistema di visione (matrice K), e non si dispone di oggetti di forma
ESTRAZIONE DI DATI 3D DA IMMAGINI DIGITALI () Calibrazione intrinseca Spesso risulta utile calibrare la sola componente intrinseca di un sistema di visione (matrice K), e non si dispone di oggetti di forma
Le proteine sono polimeri lineari costituiti da unità base formate da oltre 40 amminoacidi. Possono assumere forme diverse a seconda della funzione
 Le proteine sono polimeri lineari costituiti da unità base formate da oltre 40 amminoacidi Hanno elevato PM Possono assumere forme diverse a seconda della funzione svolgono molteplici funzioni Tra le proteine
Le proteine sono polimeri lineari costituiti da unità base formate da oltre 40 amminoacidi Hanno elevato PM Possono assumere forme diverse a seconda della funzione svolgono molteplici funzioni Tra le proteine
Relazione Laboratorio di bioinformatica
 Relazione Laboratorio di bioinformatica Davide Cittaro La predizione della struttura tridimensionale di una proteina sulla base della sequenza è un risultato ambito. L utilizzo di sole considerazioni fisico-chimiche
Relazione Laboratorio di bioinformatica Davide Cittaro La predizione della struttura tridimensionale di una proteina sulla base della sequenza è un risultato ambito. L utilizzo di sole considerazioni fisico-chimiche
Sintesi Sequenziale Sincrona Sintesi Comportamentale di reti Sequenziali Sincrone
 Sintesi Sequenziale Sincrona Sintesi Comportamentale di reti Sequenziali Sincrone Il problema dell assegnamento degli stati versione del 9/1/03 Sintesi: Assegnamento degli stati La riduzione del numero
Sintesi Sequenziale Sincrona Sintesi Comportamentale di reti Sequenziali Sincrone Il problema dell assegnamento degli stati versione del 9/1/03 Sintesi: Assegnamento degli stati La riduzione del numero
La struttura delle proteine
 La struttura delle proteine Funzioni delle proteine Strutturali Contrattili Trasporto Riserva Ormonali Enzimatiche Protezione Struttura della proteina Struttura secondaria: ripiegamento locale della catena
La struttura delle proteine Funzioni delle proteine Strutturali Contrattili Trasporto Riserva Ormonali Enzimatiche Protezione Struttura della proteina Struttura secondaria: ripiegamento locale della catena
ALLINEAMENTO DI SEQUENZE
 ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
ALLINEAMENTO DI SEQUENZE Procedura per comparare due o piu sequenze, volta a stabilire un insieme di relazioni biunivoche tra coppie di residui delle sequenze considerate che massimizzino la similarita
Come si sceglie l algoritmo di allineamento? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione?
 Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
Come si sceglie l algoritmo di allineamento? Domande: le due proteine hanno domini simili? hanno pezzi di struttura simili? appartengono alla stessa famiglia? svolgono la stessa funzione? hanno un antenato
10. Previsione della struttura tridimensionale di una proteina
 10. Previsione della struttura tridimensionale di una proteina Lo scopo di questa esercitazione è di comprendere la logica alla base della modellizzazione per omologia attraverso la previsione della struttura
10. Previsione della struttura tridimensionale di una proteina Lo scopo di questa esercitazione è di comprendere la logica alla base della modellizzazione per omologia attraverso la previsione della struttura
Alcune domande di carattere evolutivo
 Alcune domande di carattere evolutivo 1. Perché tutti gli organismi viventi (a parte le solite, rare, eccezioni usano lo stesso insieme di 20 aminoacidi? Interrelazioni tra organismi 2. Perché gli amino
Alcune domande di carattere evolutivo 1. Perché tutti gli organismi viventi (a parte le solite, rare, eccezioni usano lo stesso insieme di 20 aminoacidi? Interrelazioni tra organismi 2. Perché gli amino
Macromolecole Biologiche. Metodi di simulazione
 Metodi di simulazione Dinamica molecolare Tecnica di simulazione che permette lo studio del moto e delle proprietà di un sistema di particelle. Moti localizzati (da 0.01 a 5 Å, da 10-15 a 10-1 s) - fluttuazioni
Metodi di simulazione Dinamica molecolare Tecnica di simulazione che permette lo studio del moto e delle proprietà di un sistema di particelle. Moti localizzati (da 0.01 a 5 Å, da 10-15 a 10-1 s) - fluttuazioni
Prospettive di sviluppo
 5. DISCUSSIONE Una analisi dei risultati ottenuti con l utilizzo di SOMA e con il riferimento esterno della classificazione strutturale nota, evidenzia l insufficienza di una codifica basata sulle frequenze
5. DISCUSSIONE Una analisi dei risultati ottenuti con l utilizzo di SOMA e con il riferimento esterno della classificazione strutturale nota, evidenzia l insufficienza di una codifica basata sulle frequenze
Regressione Lineare e Regressione Logistica
 Regressione Lineare e Regressione Logistica Stefano Gualandi Università di Pavia, Dipartimento di Matematica email: twitter: blog: stefano.gualandi@unipv.it @famo2spaghi http://stegua.github.com 1 Introduzione
Regressione Lineare e Regressione Logistica Stefano Gualandi Università di Pavia, Dipartimento di Matematica email: twitter: blog: stefano.gualandi@unipv.it @famo2spaghi http://stegua.github.com 1 Introduzione
AMMINOACIDI E PROTEINE
 AMMINOACIDI E PROTEINE 1 AMMINOACIDI Gli amminoacidi sono composti organici composti da atomi di carbonio, idrogeno, ossigeno e azoto e in alcuni casi anche da altri elementi come lo zolfo. Gli amminoacidi
AMMINOACIDI E PROTEINE 1 AMMINOACIDI Gli amminoacidi sono composti organici composti da atomi di carbonio, idrogeno, ossigeno e azoto e in alcuni casi anche da altri elementi come lo zolfo. Gli amminoacidi
Metodi e Modelli per l Ottimizzazione Combinatoria Ripasso sulla Programmazione Lineare e il metodo del Simplesso (parte I)
") Metodi e Modelli per l Ottimizzazione Combinatoria Ripasso sulla Programmazione Lineare e il metodo del Simplesso (parte I) Luigi De Giovanni Giacomo Zambelli 1 Problemi di programmazione lineare Un problema
Metodi e Modelli per l Ottimizzazione Combinatoria Ripasso sulla Programmazione Lineare e il metodo del Simplesso (parte I) Luigi De Giovanni Giacomo Zambelli 1 Problemi di programmazione lineare Un problema
Immunologia e Immunologia Diagnostica MATURAZIONE DEI LINFOCITI
 Immunologia e Immunologia Diagnostica MATURAZIONE DEI LINFOCITI Il percorso di maturazione dei linfociti Sviluppo della specicifità immunologica I linfociti B e T avviano le risposte immunitarie dopo il
Immunologia e Immunologia Diagnostica MATURAZIONE DEI LINFOCITI Il percorso di maturazione dei linfociti Sviluppo della specicifità immunologica I linfociti B e T avviano le risposte immunitarie dopo il
Macromolecole Biologiche. La struttura secondaria (I)
") La struttura secondaria (I) La struttura secondaria Struttura primaria PRPLVALLDGRDETVEMPILKDVATVAFCDAQSTQEIHE Struttura secondaria La struttura secondaria Le strutture secondarie sono disposizioni regolari
La struttura secondaria (I) La struttura secondaria Struttura primaria PRPLVALLDGRDETVEMPILKDVATVAFCDAQSTQEIHE Struttura secondaria La struttura secondaria Le strutture secondarie sono disposizioni regolari
GLI ORBITALI ATOMICI
 GLI ORBITALI ATOMICI Orbitali atomici e loro rappresentazione Le funzioni d onda Ψ n che derivano dalla risoluzione dell equazione d onda e descrivono il moto degli elettroni nell atomo si dicono orbitali
GLI ORBITALI ATOMICI Orbitali atomici e loro rappresentazione Le funzioni d onda Ψ n che derivano dalla risoluzione dell equazione d onda e descrivono il moto degli elettroni nell atomo si dicono orbitali
Esercizio 1. CF 2 CS 2 CCl 4 ClF 3
 Esercizio 1 Determinare in base al metodo del legame di valenza la forma delle seguenti molecole, tenendo conto delle repulsioni coulombiane tra le coppie elettroniche di valenza CF 2 CS 2 CCl 4 ClF 3
Esercizio 1 Determinare in base al metodo del legame di valenza la forma delle seguenti molecole, tenendo conto delle repulsioni coulombiane tra le coppie elettroniche di valenza CF 2 CS 2 CCl 4 ClF 3
Correzione primo compitino, testo A
 Correzione primo compitino, testo A Parte Esercizio Facciamo riferimento alle pagine 22 e 2 del libro di testo Quando si ha a che fare con la moltiplicazione o la divisione di misure bisogna fare attenzione,
Correzione primo compitino, testo A Parte Esercizio Facciamo riferimento alle pagine 22 e 2 del libro di testo Quando si ha a che fare con la moltiplicazione o la divisione di misure bisogna fare attenzione,
Lezione 7. Allineamento di sequenze biologiche
 Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
Lezione 7 Allineamento di sequenze biologiche Allineamento di sequenze Determinare la similarità e dedurre l omologia Allineare Definire il numero di passi necessari per trasformare una sequenza nell altra
6.5 RNA Secondary Structure. 18 novembre 2014
 6.5 RNA Secondary Structure 18 novembre 2014 Calendario Oggi è la lezione 17/24: ultima lezione su Programmazione dinamica Metodo greedy: 18, 19 Grafi: 20, 21, 22, 23 Reti di flusso: 23, 24 (=mercoledì
6.5 RNA Secondary Structure 18 novembre 2014 Calendario Oggi è la lezione 17/24: ultima lezione su Programmazione dinamica Metodo greedy: 18, 19 Grafi: 20, 21, 22, 23 Reti di flusso: 23, 24 (=mercoledì
L A B C di R. Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010
 L A B C di R 0 20 40 60 80 100 2 3 4 5 6 7 8 Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010 La scelta del test statistico giusto La scelta della analisi
L A B C di R 0 20 40 60 80 100 2 3 4 5 6 7 8 Stefano Leonardi c Dipartimento di Scienze Ambientali Università di Parma Parma, 9 febbraio 2010 La scelta del test statistico giusto La scelta della analisi
Simulazione dei dati
 Simulazione dei dati Scopo della simulazione Fasi della simulazione Generazione di numeri casuali Esempi Simulazione con Montecarlo 0 Scopo della simulazione Le distribuzioni di riferimento usate per determinare
Simulazione dei dati Scopo della simulazione Fasi della simulazione Generazione di numeri casuali Esempi Simulazione con Montecarlo 0 Scopo della simulazione Le distribuzioni di riferimento usate per determinare
Il problema del commesso viaggiatore: da Ulisse alla Logistica integrata. Luca Bertazzi
 Il problema del commesso viaggiatore: da Ulisse alla Logistica integrata Luca Bertazzi 0 3 Ulisse: da Troia a Itaca Troia Itaca 509 km Quale è stato invece il viaggio di Ulisse? Il viaggio di Ulisse Troia
Il problema del commesso viaggiatore: da Ulisse alla Logistica integrata Luca Bertazzi 0 3 Ulisse: da Troia a Itaca Troia Itaca 509 km Quale è stato invece il viaggio di Ulisse? Il viaggio di Ulisse Troia
Introduzione alla programmazione lineare
 Introduzione alla programmazione lineare struttura del problema di PL forme equivalenti rappresentazione e soluzione grafica rif. Fi 1.2; BT 1.1, 1.4 Problema di programmazione lineare Dati: un vettore
Introduzione alla programmazione lineare struttura del problema di PL forme equivalenti rappresentazione e soluzione grafica rif. Fi 1.2; BT 1.1, 1.4 Problema di programmazione lineare Dati: un vettore
Corso di Bioinformatica. Docente: Dr. Antinisca DI MARCO
 Corso di Bioinformatica Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@univaq.it Analisi Filogenetica Gene Ancestrale duplicazione genica La filogenesi è lo studio delle relazioni evolutive tra
Corso di Bioinformatica Docente: Dr. Antinisca DI MARCO Email: antinisca.dimarco@univaq.it Analisi Filogenetica Gene Ancestrale duplicazione genica La filogenesi è lo studio delle relazioni evolutive tra
Stima della qualità dei classificatori per l analisi dei dati biomolecolari
 Stima della qualità dei classificatori per l analisi dei dati biomolecolari Giorgio Valentini e-mail: valentini@dsi.unimi.it Rischio atteso e rischio empirico L` apprendimento di una funzione non nota
Stima della qualità dei classificatori per l analisi dei dati biomolecolari Giorgio Valentini e-mail: valentini@dsi.unimi.it Rischio atteso e rischio empirico L` apprendimento di una funzione non nota
Omologia di sequenze: allineamento e ricerca
 Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Omologia di sequenze: allineamento e ricerca Genomi (organismi) e geni hanno un evoluzione divergente Sequenze imparentate per evoluzione divergente sono omologhe Le sequenze sono confrontabili tramite
Alberi filogenetici. File: alberi_filogenetici.odp. Riccardo Percudani 02/03/04
 Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
Alberi filogenetici The tree of life Albero filogenetico costruito con le sequenze della subunità piccola dell RNA ribosomale. Tutte le forme viventi condividono un comune ancestore (LCA, last common ancestor
RETI DI CALCOLATORI II
 RETI DI CALCOLATORI II Facoltà di Ingegneria Università degli Studi di Udine Ing. DANIELE DE CANEVA a.a. 2009/2010 ARGOMENTI DELLA LEZIONE TEORIA DEL ROUTING ROUTING STATICO ROUTING DINAMICO o PROTOCOLLI
RETI DI CALCOLATORI II Facoltà di Ingegneria Università degli Studi di Udine Ing. DANIELE DE CANEVA a.a. 2009/2010 ARGOMENTI DELLA LEZIONE TEORIA DEL ROUTING ROUTING STATICO ROUTING DINAMICO o PROTOCOLLI
Rappresentazione dei Dati Biologici
 Rappresentazione dei Dati Biologici CORSO DI BIOINFORMATICA C.d.L. Ingegneria Informatica e Biomedica Outline Proteine ed Amminoacidi Rappresentazione di Amminoacidi Rappresentazione delle strutture Proteiche
Rappresentazione dei Dati Biologici CORSO DI BIOINFORMATICA C.d.L. Ingegneria Informatica e Biomedica Outline Proteine ed Amminoacidi Rappresentazione di Amminoacidi Rappresentazione delle strutture Proteiche
Sub-Optimal Measurement-Based CAC Algorithm
 Tecniche per la garanzia di qualità in reti di Telecomunicazioni multiservizi Sottotema Controllo Courmayeur, 12-14 Gennaio 2000 Sub-Optimal Measurement-Based CAC Algorithm Gregorio Procissi procissi@iet.unipi.it
Tecniche per la garanzia di qualità in reti di Telecomunicazioni multiservizi Sottotema Controllo Courmayeur, 12-14 Gennaio 2000 Sub-Optimal Measurement-Based CAC Algorithm Gregorio Procissi procissi@iet.unipi.it
scaricato da www.sunhope.it Proteine semplici costituite dai soli amminoacidi
 Proteine semplici costituite dai soli amminoacidi Proteine coniugate costituite dagli amminoacidi + porzioni di natura non amminoacidica dette GRUPPI PROSTETICI Le Proteine coniugate prive del gruppo prostetico
Proteine semplici costituite dai soli amminoacidi Proteine coniugate costituite dagli amminoacidi + porzioni di natura non amminoacidica dette GRUPPI PROSTETICI Le Proteine coniugate prive del gruppo prostetico
Algoritmi e Strutture Dati
 Algoritmi Golosi (Greedy) Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino un algoritmo goloso correttezza Problema della selezione di attività
Algoritmi Golosi (Greedy) Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino un algoritmo goloso correttezza Problema della selezione di attività
LE PROTEINE. SONO Polimeri formati dall unione di AMMINOACIDI (AA) Rende diversi i 20 AA l uno dall altro UN ATOMO DI C AL CENTRO
 Rende diversi i 20 AA l uno dall altro UN ATOMO DI C AL CENTRO") LE PROTEINE SONO Polimeri formati dall unione di ATOMI DI C, H, N, O CHE SONO AMMINOACIDI (AA) Uniti tra loro dal Legame peptidico 20 TIPI DIVERSI MA HANNO STESSA STRUTTURA GENERALE CON Catene peptidiche
LE PROTEINE SONO Polimeri formati dall unione di ATOMI DI C, H, N, O CHE SONO AMMINOACIDI (AA) Uniti tra loro dal Legame peptidico 20 TIPI DIVERSI MA HANNO STESSA STRUTTURA GENERALE CON Catene peptidiche
Modellazione di sistemi ingegneristici (parte 2 di 2)
") Corso di Teoria dei Sistemi Modellazione di sistemi ingegneristici (parte 2 di 2) Prof. Ing. Daniele Testi DESTeC, Dipartimento di Ingegneria dell Energia, dei Sistemi, del Territorio e delle Costruzioni
Corso di Teoria dei Sistemi Modellazione di sistemi ingegneristici (parte 2 di 2) Prof. Ing. Daniele Testi DESTeC, Dipartimento di Ingegneria dell Energia, dei Sistemi, del Territorio e delle Costruzioni
Algoritmi e Strutture Dati
 Algoritmi Ricorsivi e Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino A.A. 2006/07 I conigli di Fibonacci Ricerca Binaria L isola dei conigli
Algoritmi Ricorsivi e Maria Rita Di Berardini, Emanuela Merelli 1 1 Dipartimento di Matematica e Informatica Università di Camerino A.A. 2006/07 I conigli di Fibonacci Ricerca Binaria L isola dei conigli
STRUTTURA E FUNZIONE DELLE PROTEINE
 STRUTTURA E FUNZIONE DELLE PROTEINE PROTEINE 50% DEL PESO SECCO DI UNA CELLULA STRUTTURA intelaiatura citoscheletrica strutture cellulari impalcatura di sostegno extracellulare FUNZIONE catalisi enzimatica
STRUTTURA E FUNZIONE DELLE PROTEINE PROTEINE 50% DEL PESO SECCO DI UNA CELLULA STRUTTURA intelaiatura citoscheletrica strutture cellulari impalcatura di sostegno extracellulare FUNZIONE catalisi enzimatica
Ottimizzazione Combinatoria e Reti (a.a. 2007/08)
") o Appello 6/07/008 Ottimizzazione Combinatoria e Reti (a.a. 007/08) Nome Cognome: Matricola: ) Dopo avere finalmente superato l esame di Ricerca Operativa, Tommaso è pronto per partire in vacanza. Tommaso
o Appello 6/07/008 Ottimizzazione Combinatoria e Reti (a.a. 007/08) Nome Cognome: Matricola: ) Dopo avere finalmente superato l esame di Ricerca Operativa, Tommaso è pronto per partire in vacanza. Tommaso
Regole associative Mario Guarracino Laboratorio di Sistemi Informativi Aziendali a.a. 2006/2007
 Regole associative Mario Guarracino Laboratorio di Sistemi Informativi Aziendali a.a. 26/27 Introduzione Le regole associative si collocano tra i metodi di apprendimento non supervisionato e sono volte
Regole associative Mario Guarracino Laboratorio di Sistemi Informativi Aziendali a.a. 26/27 Introduzione Le regole associative si collocano tra i metodi di apprendimento non supervisionato e sono volte
La ricerca di similarità: i metodi
 La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
La ricerca di similarità: i metodi Pairwise alignment allineamenti a coppie 1. Analisi della matrice a punti (dot matrix) 2. Programmazione dinamica (dynamic programming) allineamenti locale e globale.
Fisica Quantistica III Esercizi Natale 2009
 Fisica Quantistica III Esercizi Natale 009 Philip G. Ratcliffe (philip.ratcliffe@uninsubria.it) Dipartimento di Fisica e Matematica Università degli Studi dell Insubria in Como via Valleggio 11, 100 Como
Fisica Quantistica III Esercizi Natale 009 Philip G. Ratcliffe (philip.ratcliffe@uninsubria.it) Dipartimento di Fisica e Matematica Università degli Studi dell Insubria in Como via Valleggio 11, 100 Como
Ricerca Operativa A.A. 2007/2008
 Ricerca Operativa A.A. 2007/2008 9. Cenni su euristiche e metaeuristiche per ottimizzazione combinatoria Motivazioni L applicazione di metodi esatti non è sempre possibile a causa della complessità del
Ricerca Operativa A.A. 2007/2008 9. Cenni su euristiche e metaeuristiche per ottimizzazione combinatoria Motivazioni L applicazione di metodi esatti non è sempre possibile a causa della complessità del
Lezione 6. Analisi di sequenze biologiche e ricerche in database
 Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Lezione 6 Analisi di sequenze biologiche e ricerche in database Schema della lezione Allinemento: definizioni Allineamento di due sequenze Ricerca di singola sequenza in banche dati (Alignment-based database
Analisi degli Errori di Misura. 08/04/2009 G.Sirri
 Analisi degli Errori di Misura 08/04/2009 G.Sirri 1 Misure di grandezze fisiche La misura di una grandezza fisica è descrivibile tramite tre elementi: valore più probabile; incertezza (o errore ) ossia
Analisi degli Errori di Misura 08/04/2009 G.Sirri 1 Misure di grandezze fisiche La misura di una grandezza fisica è descrivibile tramite tre elementi: valore più probabile; incertezza (o errore ) ossia
Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation
 Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation Dipartimento di Elettronica e Informazione Politecnico di Milano Evolutionary Computation Raggruppa modelli di calcolo
Ingegneria della Conoscenza e Sistemi Esperti Lezione 9: Evolutionary Computation Dipartimento di Elettronica e Informazione Politecnico di Milano Evolutionary Computation Raggruppa modelli di calcolo
Corso di Geometria BIAR, BSIR Esercizi 2: soluzioni
 Corso di Geometria 2- BIAR, BSIR Esercizi 2: soluzioni Esercizio Calcolare il determinante della matrice 2 3 : 3 2 a) con lo sviluppo lungo la prima riga, b) con lo sviluppo lungo la terza colonna, c)
Corso di Geometria 2- BIAR, BSIR Esercizi 2: soluzioni Esercizio Calcolare il determinante della matrice 2 3 : 3 2 a) con lo sviluppo lungo la prima riga, b) con lo sviluppo lungo la terza colonna, c)
Valutazione della capacità dissipativa di un sistema strutturale
 Tecniche innovative per l identificazione delle caratteristiche dinamiche delle strutture e del danno Valutazione della capacità dissipativa di un sistema strutturale Prof. Ing. Felice Carlo PONZO - Ing.
Tecniche innovative per l identificazione delle caratteristiche dinamiche delle strutture e del danno Valutazione della capacità dissipativa di un sistema strutturale Prof. Ing. Felice Carlo PONZO - Ing.
Principi e Metodologie della Progettazione Meccanica
 Principi e Metodologie della Progettazione Meccanica ing. F. Campana a.a. 06-07 Lezione 11: CAE e Ottimizzazione Strutturale Il ruolo dell ottimizzazione nell ambito della progettazione meccanica Durante
Principi e Metodologie della Progettazione Meccanica ing. F. Campana a.a. 06-07 Lezione 11: CAE e Ottimizzazione Strutturale Il ruolo dell ottimizzazione nell ambito della progettazione meccanica Durante
Macromolecole Biologiche Interazioni non covalenti
 Interazioni non covalenti D H A δ - δ + δ - Le interazioni non covalenti Interazioni fra atomi che non sono legati da legami covalenti. Le interazioni non covalenti sono molto meno intense rispetto alle
Interazioni non covalenti D H A δ - δ + δ - Le interazioni non covalenti Interazioni fra atomi che non sono legati da legami covalenti. Le interazioni non covalenti sono molto meno intense rispetto alle
Serie storiche Mario Guarracino Laboratorio di Sistemi Informativi Aziendali a.a. 2006/2007
 Serie storiche Introduzione Per alcuni dataset, l attributo target è soggetto ad un evoluzione temporale e risulta associato ad istanti di tempo successivi. I modelli di analisi delle serie storiche si
Serie storiche Introduzione Per alcuni dataset, l attributo target è soggetto ad un evoluzione temporale e risulta associato ad istanti di tempo successivi. I modelli di analisi delle serie storiche si
SISTEMI INFORMATIVI GEOGRAFICI (GIS)
") SISTEMI INFORMATIVI GEOGRAFICI (GIS) Prof. Dipartimento di Elettronica e Informazione Politecnico di Milano SISTEMA INFORMATIVO GEOGRAFICO E UN SISTEMA CHE USA SIA DATI SPAZIALI (CIOE BASATI SU RIFERIMENTI
SISTEMI INFORMATIVI GEOGRAFICI (GIS) Prof. Dipartimento di Elettronica e Informazione Politecnico di Milano SISTEMA INFORMATIVO GEOGRAFICO E UN SISTEMA CHE USA SIA DATI SPAZIALI (CIOE BASATI SU RIFERIMENTI
Acidi Nucleici: DNA = acido deossiribonucleico
 Acidi Nucleici: DNA = acido deossiribonucleico depositario dell informazione genetica RNA: acido ribonucleico trascrizione e traduzione dell informazione genetica dogma centrale della biologia molecolare
Acidi Nucleici: DNA = acido deossiribonucleico depositario dell informazione genetica RNA: acido ribonucleico trascrizione e traduzione dell informazione genetica dogma centrale della biologia molecolare
Chimotripsina Una proteina globulare. Glicina Un amminoacido
 Chimotripsina Una proteina globulare Glicina Un amminoacido - In teoria un numero enorme di differenti catene polipeptidiche potrebbe essere sintetizzato con i 20 amminoacidi standard. 20 4 = 160.000 differenti
Chimotripsina Una proteina globulare Glicina Un amminoacido - In teoria un numero enorme di differenti catene polipeptidiche potrebbe essere sintetizzato con i 20 amminoacidi standard. 20 4 = 160.000 differenti
Allineamento e similarità di sequenze
 Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità
Allineamento e similarità di sequenze Allineamento di Sequenze L allineamento tra due o più sequenza può aiutare a trovare regioni simili per le quali si può supporre svolgano la stessa funzione; La similarità