Inquadramento clinico e diagnostico delle malattie muscolari
|
|
|
- Aniello Scala
- 10 anni fa
- Просмотров:
Транскрипт
1 Inquadramento clinico e diagnostico delle malattie muscolari

2 MIOPATIE Le miopatie sono un gruppo di malattie geneticamente determinate che si caratterizzano clinicamente per una debolezza muscolare progressiva che coinvolge i muscoli dei cingoli scapolare e pelvico e spesso il muscolo cardiaco.

3 DIAGNOSI MOLECOLARE nelle MALATTIE MUSCOLARI Limb girdle dystrophies: Dominant 1A: Myotilin; 5q31; Dysarthria 1B: Lamin A/C; 1q21; + Cardiac 1C: Caveolin-3; 3p25; Child onset 1D: 7q Dilated Cardiomyopathy (?1E): 6q23 1F: 7q32 1G: 4p21 Ankle contractures & High CK Bethlem: Collagen VI; 21q22 & 2q37 Central core: Ryanodine receptor (19q13) Cytoplasmic body: 2q24; 2q21 + Other Distal myopathies MPD2: 5q31;? Emery-Dreifuss: Lamin A/C; 1q21 Facioscapulohumeral: 4q35 Myofibrillar (Desmin storage) Desmin: 2q35; AD or AR αb-crystallin: 11q22 Filamin C: 7q32 LGMD 1A: Myotilin; 5q31 Congenital: SEPN1; 1p36 ZASP myopathy: 10q22 Other Myotonic (DM1): DMPK; 19q13 Myotonic (DM2): ZNF9; 3q21 Oculopharyngeal: PABP2; 14q11 Skeletal + Myopathy Bone fragility: 9p21 Paget disease: VCP; 9p13 Limb girdle dystrophies: Recessive 2A: Calpain-3 ;15q15 2B: Dysferlin; 2p13.1 2C: γ-sarcoglycan; 13q12 2D: α-sarcoglycan; 17q21 2E: β-sarcoglycan; 4q12 2F: δ-sarcoglycan; 5q33 2G: Telethonin; 17q H: TRIM32; 9q31-q33 2I: FKRP; 19q13.3 2J: Titin; 2q31 2K: POMT1; 9q34 Merosin (Laminin α2) Absent: 6q2 Reduced: 6q2 Abnormal: LGMD 2I Caveolin-3 mutation (Gly55Ser) Limb girdle dystrophies: X- linked Barth: G4.5 (Tafazzins); Xp28 Becker: Dystrophin; Xp21 Duchenne: Dystrophin; Xp21 Emery-Dreifuss: Emerin; Xq28 Manifesting carriers Dystrophinopathy Myotubularin McLeod Syndrome: XK; Xp21.1; Vacuolar Danon's disease: LAMP-2; Xq24 Excessive Autophagy: Xq28 Mental retardation & Cardiomyopathy Other inherited myopathy syndromes Barnes's myopathy Cardiac + Myopathy Cardiomyopathy-associated myopathy Cardiomyopathy (?LGMD1B) Dilated Cardiomyopathy: 6q23 Congenital Myopathies: Late-onset Muscular dystrophies Cytoplasmic body myopathies Distal myopathies Excessive autophagy: Xq28 Familial myasthenia gravis FSH dystrophy: 4q35 Hereditary IBM syndromes IBM1: Dominant IBM2: GNE; 9p12; Recessive IBM3: MyHC-IIa; 17p13; Dominant IBM + Paget disease: 9p13; Dominant Metabolic myopathies Glycogen Lipid Mitochondrial Myopathy + PEO: 17p13; Recessive Myotonic dystrophy Other dystrophies Respiratory failure Scapuloperoneal syndromes Skeletal + Myopathy Diaphyseal dysplasia: TGFB1; 19q13 Epiphyseal dysplasia: COL9A3; 20q13 Spheroid body (Myotilin) Tubular aggregates Tubular arrays
 Desmin: 2q35; AD or AR αb-crystallin: 11q22 Filamin C: 7q32 LGMD 1A: Myotilin; 5q31 Congenital: SEPN1; 1p36")
4 DISTROFIE MUSCOLARI Le distrofie muscolari sono un gruppo di malattie geneticamente determinate che si caraterizzano clinicamente per una debolezza muscolare progressiva che coinvolge i muscoli dei cingoli scapolare e pelvico e spesso il muscolo cardiaco. Il muscolo scheletrico mostra dal punto di vista istopatologico una progressiva sostituzione fibro-adiposa, degenerazione e rigenerazione (DISTROFIA).

5 Muscolo normale Muscolo distrofico H&E

6 LA BIOPSIA MUSCOLARE Selezione del muscolo appropriato Procedura bioptica Processamento del campione

7 Criteri di selezione del muscolo da biopsiare Evitare muscoli troppo deboli (i.e. ampiamente sostituiti da tessuto fibro-adiposo) o muscoli completamente normali (i.e. nessuna alterazione patologica) Scegliere un muscolo con debolezza di grado moderato (3+-4 MRC) Il muscolo deve essere esente da recenti traumi (EMG) o da patologia concomitante Tricipite, bicipite all arto superiore; quadricipite all arto inferiore Indicazioni particolari

8

9 PREPARAZIONE del CAMPIONE Si preleva un frammento di dimensioni adeguate per: Congelamento Routine istopatologica Immunoistochimica Biochimica RNA Microscopia elettronica Colture cellulari Il campione per il congelamento viene congelato in isopentano raffreddato in azoto liquido e conservato in ultra-freezer a -80 C.

10 ROUTINE ISTOCHIMICA ed ISTOENZIMATICA Ematossilina ed eosina Tricromica di Gomori Enzimi ossidativi NADH-TR SDH COX ATPasi ph 4.3 ph 9.4 PAS ORO Fosfatasi acida Morfologia della fibra muscolare, elementi non contrattili del muscolo, nervi, vasi, tessuto connettivo, adiposo, nuclei, strutture anomale nelle fibre muscolari (p.es. corpi nemalinici) Colorano differenzialmente i diversi tipi di fibra e dimostrano alterazioni nella distribuzione dei mitocondri e delle miofibrille Colorano differenzialmente i diversi tipi di fibra e dimostrano alterazioni della innervazione Colora il glicogeno e gli altri polisaccaridi Colora i lipidi neutri Positiva in fibre necrotiche, o in membrane con trasporto attivo
 Colorano differenzialmente i diversi tipi di fibra e dimostrano alterazioni nella distribuzione dei mitocondri e delle miofibrille Colorano differenzialmente")
11 MIOPATIE Le miopatie sono un gruppo di malattie geneticamente determinate che si caratterizzano clinicamente per una debolezza muscolare progressiva che coinvolge i muscoli dei cingoli scapolare e pelvico e spesso il muscolo cardiaco.

12 TRASMISSIONE LEGATA AL CROMOSOMA X
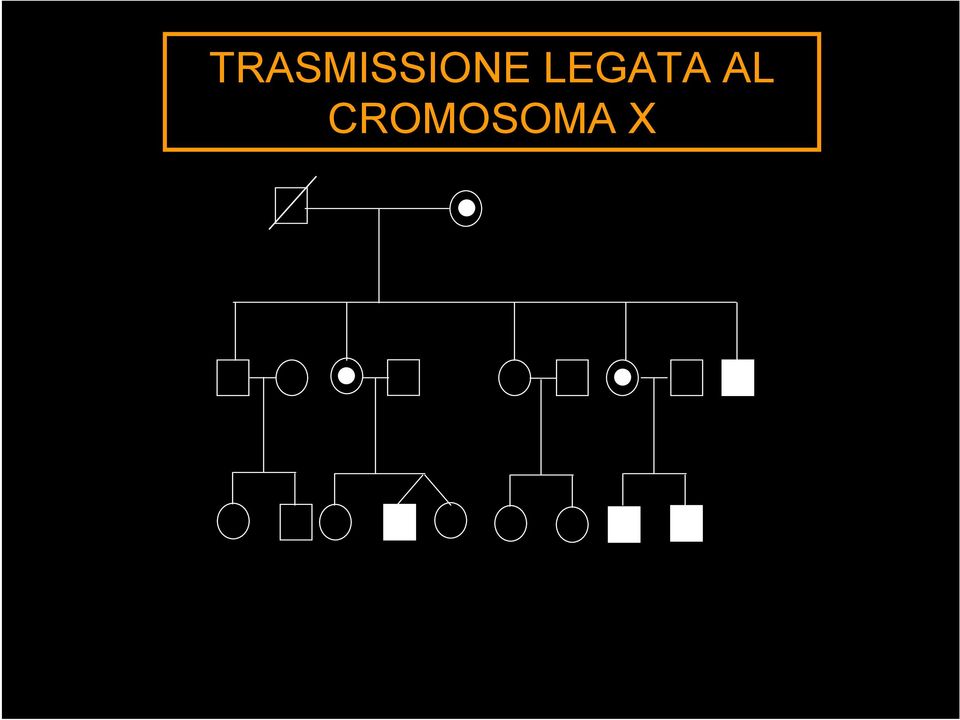
13 DISTROFINOPATIE Distrofia muscolare di Duchenne (DMD) Distrofia muscolare di Becker (BMD) Portatrice di DMD/BMD
 Portatrice di")
14 DISTROFINOPATIE Sono dovute a mutazioni nel gene DMD che mappa sul braccio corto del cromosoma X in p21 e che codifica per una proteina chiamata distrofina

15 TRASMISSIONE DI UNA MALATTIA LEGATA AL CROMOSOMA X gene DMD mutato figlia portatrice (50%) figlio affetto (50%)
")
16 DISTROFINA Il piu grande gene umano ( bp) 10 volte piu grande di qualsiasi altro gene Espressione: muscolo scheletrico muscolo liscio muscolo cardiaco alcune popolazioni neuronali Prodotto proteico: DISTROFINA (427 kda) Componente del citoscheletro Funzioni: conferisce stabilita alla membrana muscolare durante lo sforzo stabilizza le altre glicoproteine associate alla membrana Quantita : 0.002% delle proteine muscolari 3% delle proteine di membrana

17 Distrofia muscolare di Duchenne Epidemiologia,tasso di mutazione Interessa tutte le popolazioni egulmente, elevato tasso di mutazione spontanea 1/ cellule uovo o spermatozoi hanno mutazioni del gene per la distrofina de novo 1/3.500 maschi nati vivi sono colpiti da DMD

18 Distrofia muscolare di Duchenne caratteristiche cliniche Periodo neonatale nella norma Ritardata l acquisizione delle tappe motorie Esordio della debolezza muscolare prossimale verso i 2-3 anni Debolezza e/o atrofia muscolare progressiva Perdita della deambulazione verso i anni Prognosi basata sulla funzionalita respiratoria e cardiaca

19 Misurazione manuale della forza muscolare M.R.C. (Medical Research Council) Normale Movimento possibile anche contro resistenza, non massimale Movimento possibile solo contro gravita ma non contro resistenza Movimento possibile solo con gravita eliminata Contrazione muscolare senza movimento Nessuna contrazione Si assegna un punteggio da 0 a 5 per ogni muscolo testato

20 Protocollo di valutazione GSGC (Gait, Stair, Gowers, Chair) 1) MARCIA (GAIT) 1. Normale 2. Lievemente ancheggiante, in iperlordosi e/o sulle punte dei piedi 3. Moderatamente ancheggiante, in iperlordosi e/o sulle punte dei piedi 4. Severamente ancheggiante, in iperlordosi e/o sulle punte dei piedi 5. Cammino possibile solo con assistenza (p.es. stampelle, tutori di posizione, etec.) 6. Mantiene la stazione eretta, ma incapace di camminare 7. Confinato in sedia a rotelle Tempo di percorrenza di 10 metri:...sec. 2) SALIRE le SCALE (STAIRS) 1. Sale senza appoggio 2. Sale con la spinta di una mano sulla coscia 3. Sale con la spinta di entrambe le mani sulle coscie 4. Sale in posizione eretta ma con l aiuto del corrimano 5. Sale con appoggio di entrambe le mani sul corrimano 6. Sale solo pochi gradini 7. Incapace di salire le scale Tempo per salire 4 gradini:..sec. 3) DA SEDUTO AD ERETTO (GOWERS) 1. Normale 2. Solleva il bacino per primo, una mano sul pavimento 3. Solleva il bacino per primo, due mani sul pavimento 4. Si appoggia con una mano sulla coscia 5. Si appoggia con due mani sulle coscie 6. Si solleva solo con l appoggio agli oggetti vicini (tavolo, sedia, etc.) 7. Incapace di alzarsi da terra Tempo per alzarsi da terra: sec. 4) ALZARSI DA UNA SEDIA (CHAIR) 1. Normale 2. Su base allargata e/o difficolta ma senza appoggio 3. Con appoggio di una mano sulla coscia 4. Con appoggio di entrambe le mani sulle coscie 5. Con appoggio sulla sedia o oggetti vicini 6. Incapace di alzarsi Tempo per alzarsi da una sedia:..sec.

21 Distrofia muscolare di Becker caratteristiche cliniche Esordio della debolezza muscolare prossimale tra i 5-15 anni nelle forme precoci, ma anche esordi tardivi nella terza o quarta decade ed oltre Debolezza e/o atrofia muscolare progressiva Perdita della deambulazione dopo i 15 anni Fenotipo clinico estremamente eterogeneo Prognosi basata sulla funzionalita respiratoria e cardiaca
22 Polimorfismo clinico nei pazienti con BMD IperCKemia, mialgie, crampi Mioglobinuria, intolleranza all esercizio Miopatia dei quadricipiti Miopatia ad esordio tardivo Cardiomiopatia dilatativa X-linked
23 Dati ecocardigrafici in pazienti BMD
24 Correlazioni fra LVEDV e LVEF ed eta dei pazienti BMD
25 CASO ISOLATO di DEFICIT di FORZA PROGRESSIVO
26 ALGORITMO DIAGNOSTICO Anamnesi ed esame neurologico Esami generali: CPK EMG: miopatico/neurogeno BIOPSIA MUSCOLARE (immunoistochimica, immunoblotting) Analisi del DNA
27 Nei muscoli dei vertebrati la fosfocreatina costituisce una riserva di gruppi fosfato che possono esere trasferiti all ATP. fosfocreatina+ ADP + H+ ATP + creatina Enzima che catalizza la reazione = creatina chinasi
28 Prelievo di sangue Anamnesi ed esame neurologico Biopsia muscolare DNA PROTEINE DIAGNOSI
29 Analisi proteica distrofinopatie * muscolo normale distrofina normale muscolo DMD distrofina assente muscolo BMD distrofina ridotta fibre revertite (30% dei casi) C C C P C C C distrofina 427 kda miosina
30 DISTROFIA DI DUCHENNE ricerca di mutazioni nel gene per la distrofina P1 P2 C C P1 P2 C P1 deleto esone 51 P2 deleto esoni compresi 52
31 Mutazioni del gene per la distrofina Tipo di mutazione Delezione di uno o piu esoni Duplicazione di uno o piu esoni Mutazioni puntiformi DMD 65% 6-10% 25-30% BMD 85% 6-10% 5-10% 2 hotspots: uno nel dominio rod (esoni 45-55) ed uno alla estremita 5 (esoni 2-19) Lo screening di 19 esoni identifica il 98% delle delezioni
32 GENE per la DISTROFINA DP427 muscolo, neuroni quadro di lettura senza-slittamento DP260 retina DP140 neuroni, rene DP116 cellule di Schwann DP71 45 universale con slittamento
33 Mutazioni frame-shift e in-frame gene DMD sintrofina distrofina nnos α-actinina actina distrobrevina Mutazioni frame-shift Mutazioni in-frame distrofina assente C P P C P C C * * * distrofina di ridotta aaaaaaquantita e/o aaaaapeso aaaaamolecolare Fenotipo severo DMD Fenotipo mite BMD
34 DISTROFIA MUSCOLARE di Miglioramento e mantenimento della forza muscolare e funzione muscolare Prevenzione e gestione delle deformita spinali Gestione delle complicanze respiratorie Prevenzione e trattamento della cardiomiopatia DUCHENNE
35 FOLLOW-UP CLINICO nelle DISTROFINOPATIE Valutazione neurologica EKG, ecocardiogramma e visita cardiologica Spirometria, emogas analisisi e visita pneumologica Visita fisiatrica e cicli di FKT Valutazione nutrizionale
36 TRATTAMENTO delle DISTROFINOPATIE Mantenere la deambulazione (ortesi, fisiatria, interventi chirurgici) Prevenire l insufficienza respiratoria (ventilazione non invasiva) Prevenire l insufficienza cardiaca (profilassi precoce con ACE inibitori) Trattamento farmacologico
37 Distrofia muscolare di Duchenne perdita della deambulazione Age at loss of ambulation Natural history DMD (n=85) Steroid treated DMD (n=41) 7 5 3
38 PORTATRICI di DISTROFIA MUSCOLARE di DUCHENNE Il 90% delle portatrici di DMD e asintomatico Circa il 10% delle portatrici di DMD presenta segni clinici di miopatia Poche fibre distrofino-negative Molte fibre distrofino-negative
39 INATTIVAZIONE DEL CROMOSOMA X RANDOM cromosoma X con il gene per la distrofina normale attivo in ~ 50% delle cellule dys- dys+ dys- dys- dys- dys+ dys+ dys+ dys+ dys- dys- dys+ dys+ dys- dys- dys+ INATTIVAZIONE DEL CROMOSOMA X SBILANCIATA Cromosoma X con il gene per la distrofina normale in ~ 100% delle cellule dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys+ dys- dys- dys- dys- dys- dys ḏysdysdysdys- dys- dysdys- dys- dys- portatrice sintomatica Cromosoma X con il gene per la distrofina mutato in ~ 100% delle cellule
40 sospetto di distrofia muscolare biopsia muscolare IF distrofina assente mosaico ridotta normale DMD DMD carrier immunoblot distrofina ridotta normale BMD LGMD?
41 LGMD Coinvolgono prevalentemente i muscoli prossimali con risparmio dei muscoli faciali, extraoculari e faringei Ampia sovrapposizione tra i vari quadri clinici delle diverse LGMD eterogeneita clinica Mutazioni in numerosi geni causano LGMD eterogeneita genetica
42 Distrofie muscolari dei cingoli autosomiche dominanti (LGMD1) LGMD INHERITANCE LOCUS PROTEIN LGMD1A AD 5q myotilin LGMD1B AD 1q11 lamin A/C LGMD1C AD 3p caveolin LGMD1D AD 6q22 - LGMD1E AD 7q - LGMD1F AD 7q - LGMD1G AD 4p21 -
43 Distrofie muscolari dei cingoli autosomiche recessive (LGMD2) LGMD INHERITANCE LOCUS PROTEIN LGMD2A AR 15q calpain-3 LGMD2B AR 2p dysferlin LGMD2C AR 13q γ-sarcoglycan LGMD2D AR 17q α-sarcoglycan LGMD2E AR 4q β-sarcoglycan LGMD2F AR 5q δ-sarcoglycan LGMD2G AR 17q telethonin LGMD2H AR 9q TRIM32 LGMD2I AR 19q FKRP LGMD2J AR 2q31 titin LGMD2K AR 9q34 POMT1 LGMD2L AR 9q31 FKTN LGMD2M AR 1p34 POMGnT1 LGMD2N AR 14q24 POMT2
44 LGMD2D Chr. 17q12-21 LGMD2C Chr. 13q12 LGMD2E Chr. 4q12 LGMD2F Chr. 5q33-34 DMD/BMD Chr.Xp21 LGMD1C Chr.3p LGMD1B Chr.1q FKRP LGMD2B Chr.2p LGMD2A Chr.15q LGMD2I Chr 19q13.3
45 Calpainopatie (LGMD2A) Esordio clinico usualmente entro le prime decadi Coinvolgimento del cingolo scapolare all esordio (forma di Erb) o del cingolo pelvico con risparmio degli abduttori delle coscie; Ipotrofia muscolare e scapola alata frequenti; Contratture dei tendini di Achille all esordio; Insufficienza respiratoria
46 Disferlinopatie (LGMD2B) LGMD Esordio nell eta giovanile Lentamente progressiva Esordio nei muscoli prossimali del cingolo pelvico Risparmio dei muscoli della loggia anteriore della gamba e dei muscoli distali degli arti Miopatia distale (miopatia di Miyoshi) Esordio nella vita adulta Precoce coinvolgimento del gastrocnemio (impossibilita a camminare sulle punte dei piedi) Ipotrofia distale arti inferiori Frequente progressione ai muscoli prossimali ed al cingolo scapolare
47 Sarcoglicanopatie (LGMD2C-F) Esordio clinico variabile (dall infanzia alla vita adulta) Coinvolgimento del cingolo pelvico maggiore del cingolo scapolare; Ipertrofia dei polpacci comune; Variabile progressione: da molto severa (DMD-like) a molto mite; Coinvolgimento cardiaco (LGMD2E-2F) e respiratorio frequenti
48 LGMD2I Esordio clinico variabile (dall infanzia alla vita adulta) Coinvolgimento del cingolo pelvico e cingolo scapolare; Ipertrofia dei polpacci e della lingua comune; Variabile progressione nella LGMD2I: da molto severa (DMD-like) a molto mite; Cardiomiopatia dilatativa ed insufficienza respiratoria comuni nella LGMD2I
49 ISTOPATOLOGIA severa moderata mite Pattern distrofico o miopatico di variabile severita Il fenotipo clinico e la patologia muscolare sono aspecifiche CARATTERIZZAZIONE MOLECOLARE
50 Distrofie muscolari dei cingoli autosomiche dominanti (LGMD1) LGMD INHERITANCE CHROMOSOME PROTEIN LGMD1A AD 5q myotilin LGMD1B AD 1q11-21 lamin A/C LGMD1C AD 3p caveolin LGMD1D AD 6q22 - LGMD1E AD 7q - LGMD1F LGMD1G AD AD 7q 4p Distrofie muscolari dei cingoli autosomiche recessive (LGMD2) LGMD INHERITANCE CHROMOSOME PROTEIN LGMD2A AR 15q calpain-3 LGMD2B AR 2p dysferlin LGMD2C AR 13q γ-sarcoglycan LGMD2D AR 17q α-sarcoglycan LGMD2E AR 4q β-sarcoglycan LGMD2F AR 5q δ-sarcoglycan LGMD2G AR 17q telethonin LGMD2H AR 9q TRIM32 LGMD2I AR 19q FKRP LGMD2J AR 2q31 titin LGMD2K AR 9q34 POMT1 LGMD2L AR 9q31 fukutin
51 III-20 control 237 kda Dysferlin -> LGMD2B 94 kda Calpain -> LGMD2A 50 kda α-sarcoglycan -> LGMD2D myosin
52 α-sarcoglycan LGMD2D β-sarcoglycan LGMD2E γ-sarcoglycan LGMD2C telethonin LGMD2G
53 Laminin α2 α-dystroglycan Control III-20 LGMD2I/2K/2L
54 ALGORITMO DIAGNOSTICO Anamnesi ed esame neurologico Esami generali: CPK EMG: miopatico/neurogeno BIOPSIA MUSCOLARE (immunoistochimica, immunoblotting) Analisi del DNA
55 Un approccio pratico alle distrofie muscolari Storia ed esame obiettivo: Esami generali: CPK Distrofia muscolare Biopsia muscolare Distrofia/miopatia Miopatia infiammatoria Atrofia neurogena
56 BIOPSIA MUSCOLARE Analisi immunoistochimica ed immunoblotting delle proteine coinvolte nelle distrofie muscolari Analisi genetica
57 Coinvolgimento dei muscoli mimici DISTROFIA FACIO-SCAPOLO-OMERALE (FSH)
58 Distribution of predominant muscle weakness in different types of dystrophy DMD/BMD FSH LGMD Lancet 2002; 359:
59 FSHD Autosomica Dominante (10% nuove mutazioni, 45% mosaicismo germinale) Prevalenza 1: 20,000 Penetranza incompleta: 83% all eta di 30 anni (piu alta nei maschi (95%) che nelle femmine (69%) Non-penetranza: inferiore al 2% dopo i 50 anni
60 FSHD Esordio: M=16 anni F=20 in media, ma possibile dalla prima alla sesta decade Variabilita intra ed interfamiliare (nei casi familiari fino al 40% delle femmine ed al 20% dei maschi possono essere asintomatici)
61 FSHD Esordio: Debolezza muscolare a carico di: Muscoli facciali (anche asimmetricamente) Muscoli del cingolo scapolare Muscoli estensori dei piedi (piede cadente) Progressione della debolezza muscolare Muscoli addominali Muscoli estensori dei piedi Muscoli del cingolo scapolare (deltoide risparmiato) Muscoli del cingolo pelvico
62 FSHD Sintomi associati: Difetti di conduzione (p.es. blocco di branca) (5%) Scoliosi Contratture infrequenti, tranne che alla tibiotarsica Petto excavatus (5%) Perdita dei toni alti all audiogramma (75%) Vasculopatia retinica (teleangectasie e microaneurismi capillari) (60%) Sindrome di Coat rara (larghi essudati ed emorragie) (1%)
63 Vasculopatia retinica teleangectasie e microaneurismi capillari
64 FORMA INFANTILE Circa il 2-4% di tutti i casi di FSH. Esordio della debolezza facciale prima dei 5 anni (difficoltà a dormire con gli occhi completamente chiusi, difficoltà a sorridere) e della debolezza al cingolo scapolare prima dei 10 anni. Severa e diffusa debolezza che coinvolge anche il cingolo pelvico, raramente asimmetrica % dei casi perdono la deambulazione, di solito prima dei 15 anni Possibile ritardo mentale, epilessia, coinvolgimento cardiaco, sordità, vasculopatia retinica. Frammento D4Z4 di piccole dimensioni (genitori lievemente affetti o mosaici)
65 DISTROFIA FACIO-SCAPOLO-OMERALE (FSH) basi molecolari Linkage sulla regione subtelomerica del cromosoma 4 (4q35) Nella regione telomerica del cromosoma 4 e 10 è localizzata una sequenza polimorfica di DNA detta D4Z4, costituita da unità ripetute in tandem della dimensione ciascuna di 3.3 kb Il numero di repeats varia da 11 a 150 nei controlli, rendendo le dimensioni del frammento variabili da 35 a 300 kb (11 o piu D4Z4 repeats). Patologico: meno di 35 kb, meno di 11 D4Z4 repeats cen (D4Z4) n tel ( D4Z4 D4Z4 D4Z4 D4Z4 D4Z4 D4Z4 D4Z4 D4Z4 ) n cromosoma 4q35
66 DISTROFIA FACIO-SCAPOLO- OMERALE (FSH) (D4Z4) n = range normale oltre a 11 sequenze ripetute (frammenti superiori alle 50 Kb) MUTAZIONE: sequenze D4z4 inferiori a 11 (frammenti inferiori alle 38 Kb) ANTICIPAZIONE non su base genetica (frammento stabilmente trasmesso) Goto et al; Neuromusc Disord 2006; 16:
67 CORRELAZIONI GENOTIPO-FENOTIPO C è una correlazione tra l ampiezza del frammento D4Z4 e l età di esordio e di perdita di deambulazione. Solitamente, tanto più piccolo è il frammento, tanto peggiore è il fenotipo. da 1-3 repeats (<10 kb): forma severa da 4 a 7 repeats (10-23 kb): forma classica da 8 a 10 repeats (23-35 kb): forma lieve o asintomatica
68 MECCANISMI PATOGENETICI Compromissione della struttura del gene FSH localizzato nelle sequenze D4Z4 (zona ricca di CG -> homodomain gene DUX) Controllo trascrizionale di geni vicini alle sequenze D4Z4 (effetto posizionale) (FRG1, ANT1, PDLIM3) Delezione di un silenziatore trascrizionale localizzato in D4Z4 che comporta la sovraespressione nel muscolo di pazienti affetti da FSH di geni localizzati centromericamente alle sequenze D4Z4 in 4q35 (un interruttore genetico che non funziona lascia acceso un gene che normalmente dovrebbe restare spento).
69 MIOTONIE
70 MIOTONIA: ALTERATO RILASCIAMENTO MUSCOLARE DOPO CONTRAZIONE MUSCOLARE PROTRATTA
71 Electromyography Myotonia A WT Mutant B 1 mv 0.5 s Courtesy of Lawrence J. Hayward 1 mv 0.1 s
72 Myotonic Signs Eyelid myotonia after squinting Lid lag myotonia after sustained upgaze Percussion myotonia
73 Classificazione delle miotonie Miotonina protein kinasi (DM1) Distrofia miotonica tipo 1 (AD) Zinc finger protein 9 (DM2) Distrofia miotonica tipo 2 (DM2) (AD) Canale del cloro Miotonia congenita di Thomsen (AD) Miotonia congenita di Becker (AR) Canale del sodio Paramiotonia congenita Miotonia aggravata dal potassio
74 MIOTONIA Distrofia miotonica (DM1) Distrofia miotonica (DM2) Miotonia Congenita Miotonia di Becker Miotonia aggravata dal Potassio Paramiotonia Congenita DMPK ZNF9 Canale del Cl - (CLCN1) Canale del Cl - (CLCN1) Canale del Na + (SCN4A) Canale del Na + (SCN4A)
75 EPIDEMIOLOGIA delle DM DM1 Incidenza: 13 : 100,000 Prevalenza: 2-5 / 100,000
76 DISTROFIA MIOTONICA tipo 1 o Distrofia miotonica di Steinert Pleiotropismo coinivolgimento di piu organi
77 DISTROFIA MIOTONICA (DM1) Miotonia (alterato rilasciamento muscolare) Debolezza muscolare e atrofia Muscoli mimici, temporali, muscoli distali dell avambraccio, muscoli dorsiflessori del piede Cataratta Calvizie precoci Cardiomiopatia (difetti di conduzione, iniziali; cardiomiopatia dilatativa tardiva) Atrofia gonadica, altri disturbi endocrinologici Deficit cognitivo fino alla demenza
78 DISTROFIA MIOTONICA di TIPO 1 Christmas tree cataract
79 PROFILO NEUROCOGNITIVO La popolazione DM1 è estremamente eterogenea. Nel complesso non si evidenzia un significativo deficit cognitivo, con QI medio ancora nella norma. I singoli test documentano: deficit di funzione frontale; deficit di abilità visuo-spaziali.
80 DISTROFIA MIOTONICA CONGENITA Trasmissione per via materna (la madre ha spesso un fenotipo mite o subclinico -> anticipazione) Assenza di miotonia nelle forme congenite (puo comparire dopo i 2-3 anni) Caratteristiche cliniche pre-, post-natali Idrope fetale Ipotonia Distress respiratorio Debolezza muscoli facciali Difficolta nella deglutizione Piede torto e/o contratture multiple Caratteristiche cliniche durante l infanzia Dismorfia facciale Ritardo tappe psico-motorie Ritardo nell acquisizione del linguaggio Ritardo mentale
81 DISTROFIA MIOTONICA: 19q13.3 Gene responsabile: DMPK Proteina: DMPK (serin-treonin kinasi) (autosomica dominante) ATG TAG (CTG) n 5 UTR 3 UTR 14 kb gene; 2.3 kb mrna
82 DISTROFIA MIOTONICA (DM1) (CTG) n = range normale 3-37; Eterozigosita : 0.75% MUTAZIONE > 50 (CTG)n instabilita meiotica e mitotica normale DM1 ANTICIPAZIONE Classe E1 = (CTG) n Classe E2 = 200-1,000 (CTG) n mosaico somatico Classe E3 = > 1,000 (CTG) n * ** * * * * * * Bonifazi, E. et al. Clin Chem 2006;52: ,300(CTG) n 80 (CTG) n Diagn Mol Pathol : Am J Med Genet 1996; 65:
83 DISTROFIA MIOTONICA di TIPO 1 (DM1) Fenomeno dell anticipazione
84 DM1: IPOTESI PATOGENETICHE Effetto dominante negativo dell RNA: i trascritti che contengono la sequenza ripetuta CUG si accumulano come inclusioni ribonucleiche nel nucleo (foci di RNA) Alterazione dell attivita di numerose proteine che legano l RNA (RNA binding protein) Splicing alternativo e funzione anomala di numerosi geni target L effetto tossico è proporzionale all entità dell espansione: più mrna-cug più tossicità
85 Iter diagnostico EMG: miopatia e miotonia ECG: difetti di conduzione Biopsia muscolare: ipotrofia di fibre di tipo I con nuclei centrali e masse sarcoplasmatiche o aggregati tubulari Genetica: malattia autosomica dominante con variabilita clinica
86 ISTOPATOLOGIA DM1 H&E: Nuclei centrali e polidimensionalismo fibrale. SDH: Ipotrofia delle fibre di tipo I (scure).
87 ISTOPATOLOGIA DM1 H&E H&E Polidimensionalismo fibrale Nuclei centrali Fissurazioni citoplasmatiche Aggregati tubulari NADH
88 DISTROFIA MIOTONICA tipo 2 o PROMM Pleiotropismo coinivolgimento di piu organi
89 DISTROFIA MIOTONICA di TIPO 2 DM2 mappa su 3q21; causata da una espansione di una (CCTG)n nel primo introne della proteina zinc finger 9 (ZNF9)
90 DISTROFIA MIOTONICA (DM2) Miotonia (alterato rilasciamento muscolare) Debolezza muscolare prossimo>distale e ipertrofia dei polpacci Ipotrofia associata a miotonia Cataratta Cardiomiopatia (difetti di conduzione, aritmie; cardiomiopatia dilatativa) Atrofia gonadica, altri disturbi endocrinologici Ridotta tendenza all anticipazione Elevata eterogeneita somatica ATG TAG (CCTG) n 5 UTR 3 UTR 11.3 kb gene; 1.5 kb mrna Gene ZNF9; cromosoma 3q21.3
91 DISTROFIA MIOTONICA (DM2) (TG) n (TGTG) n (CCTG) n = range normale fino a 26 MUTAZIONE (CCTG)n : da 75 a 11,000 (CCTG)n (in media circa 5,000) instabilita somatica e intergenerazionale Eterogeneita somatica Eterogeneita in gemelli Eterogeneita nel tempo DM2 controllo DM1 C. L. Liquori et al., Science 293, (2001) C. L. Liquori et al., Science 293, (2001)
92 ISTOPATOLOGIA DM2 H&E: Polidimensionalismo fibrale e rari nuclei centrali ATPasi 9.4: Atrofia prevalente delle fibre di tipo II SDH: Agglomerati di nuclei picnotici
93 DM1 DM2 Distribuzione ipostenia facciale, extraoculare ++ + distale agli arti ++ + prossimale agli arti + ++ atrofia muscolare ++ + Dolore muscolare - + sistema nervoso centrale ritardo mentale + - ipersonnia ++ - atrofia cerebrale + - lesioni della sostanza bianca +? malattie congenite + - anticipazione genetica ++ -
94 MIOTONIA CONGENITA
95 MIOTONIA Distrofia miotonica (DM1) Distrofia miotonica (DM2) Miotonia Congenita Miotonia di Becker Miotonia aggravata dal Potassio Paramiotonia Congenita DMPK ZNF9 Canale del Cl - (CLCN1) Canale del Cl - (CLCN1) Canale del Na + (SCN4A) Canale del Na + (SCN4A)
96 Canale del cloro muscolo scheletrico voltaggio-dipendente CLCN1
97 Canale del cloro (CLCN1) Ridotta conduttanza al cloro nelle cellule muscolari ipereccitabilita della membrana (alterata ripolarizzazione) Il canale del cloro e un omodimero. Ciascuna subunita forma un poro. Le mutazioni recessive agiscono con un meccanismo di loss of function, mentre le mutazioni dominanti agiscono con effetto dominant-negative
98 MIOTONIE CONGENITE Caratterizzate da senso di rigidita muscolare prevalente a carico degli arti inferiori Miotonia piu evidente quando il movimento massimale avviene dopo il riposo Fenomeno del warming-up (rigidita muscolare che migliora dopo un esercizio ripetuto) Ipertrofia muscolare Nelle forme recessive (miotonia di Becker) l esordio e piu tardivo, la miotonia e piu grave e si possono associare a miopatia tardiva
99 Clinical Diagnosis of The Skeletal Muscle Chloride Ion Channelopathies Thomsen s Disease Becker s Myotonia Inheritance autosomal dominant (AD) autosomal recessive (AR) Genetic defect loss-of-function and dominant-negative mutations in the skeletal muscle voltage-gated chloride channel gene (CLCN1 Chr 7q35) Age of onset infancy to early childhood late childhood to early teens Areas affected legs and hands most muscles including facial muscles Muscle hypertrophy legs and trunk legs Lid lag yes yes Grip myotonia yes yes Percussion myotonia yes yes EMG myotonia yes yes Muscle weakness no yes Severity minimal to mildly affected moderate to severely affected Provocative stimuli prolonged rest, initiation of movement, or maintenance of the same posture Warm-up phenomenon yes yes Therapy mexiletine, quinine, procainamide, phenytoin
100 Canale del sodio voltaggiodipendente muscolo scheletrico SCN4A
101 Membrane Topology of Voltage-Gated Sodium Channels The sodium channel Nav1.4 is made up of a principal pore forming and voltage sensitive subunit (α subunit) associated with an accessory β1 subunit. Mutations in sodium channel diseases are in the α subunit of Nav 1.4 encoded by the gene SCN4A located on chromosome 17q23-25 P-loop Inactivation gate Frances M. Ashcroft. Ion Channels and Disease. Academic Press. 2000: 70
102 Gating Properties of Voltage-Sensitive Sodium Channel Louis Ptacek. Am J Med 104:62, 1998 SCN4A mutations cause a disruption of fast inactivation (gain of function mutations) Channel re-openings and Na+ accumulation Depolarization of muscle cells and generation of repetitive action potentials Mild depolarization long lasting hyperexcitability MYOTONIA Strong depolarization general opening of the the Na+ channel PARALYSIS
103 Paramiotonia Congenita Von Eulenburg A (1886) Über eine familiare, durch 6 generationen verfolgbare form congenitaler paramyotonie. Neurologisches Centralblatt 12: Autosomica dominante con elevata penetranza Esordio infantile Miotonia paradossa Stifness muscolare indotta dal freddo seguita da debolezza muscolare/paralisi
104 Clinical Diagnosis of The Skeletal Muscle Sodium Ion Channelopathies Paramyotonia Congenita (PC, PMC) Potassium-Aggravated Myotonia (PAM) Inheritance autosomal dominant (AD) autosomal dominant (AD) Genetic defect missense mutations in the skeletal muscle voltage-gated sodium channel a-subunit gene (SCN4A Chr 17q23-25) Age of onset infancy to early childhood early childhood to second decade Areas affected facial, pharyngeal, distal facial muscles, limbs upper extremity muscles Attack duration variable variable Paralysis severity usually mild never Myotonia severity mild to moderate mild to severe Myopathy very rare rare Provocative stimuli cold, exercise, oral potassium oral potassium oral potassium, rest after exercise Therapy acetazolamide, mexiletine, acetazolamide, mexiletine tocainide, hydrochlorothiazide Animal model N/A N/A Note myotonia fluctuans myotonia permanens acetazolamide-responsive myotonia painful myotonia
distrofia dys = cattivo trophía = nutrimento
 DISTROFIA MUSCOLARE distrofia dys = cattivo trophía = nutrimento M Disco Z SARCOMERO Banda M Disco Z Unità strutturale e funzionale del muscolo striato Disco Z Nebuline COSTAMERI COSTAMERI GIUNZIONE
DISTROFIA MUSCOLARE distrofia dys = cattivo trophía = nutrimento M Disco Z SARCOMERO Banda M Disco Z Unità strutturale e funzionale del muscolo striato Disco Z Nebuline COSTAMERI COSTAMERI GIUNZIONE
INQUADRAMENTO CLINICO delle MIOPATIE
 INQUADRAMENTO CLINICO delle MIOPATIE Miopatie il danno e primitivamente muscolare Atrofie neurogene il danno muscolare e secondario ereditarie acquisite acute croniche Miopatie Distrofie muscolari Miopatie
INQUADRAMENTO CLINICO delle MIOPATIE Miopatie il danno e primitivamente muscolare Atrofie neurogene il danno muscolare e secondario ereditarie acquisite acute croniche Miopatie Distrofie muscolari Miopatie
Malattie del muscolo scheletrico da primitiva compromissione strutturale e/o funzionale. Spesso causa di grave disabilità
 MIOPATIE Malattie del muscolo scheletrico da primitiva compromissione strutturale e/o funzionale. Spesso causa di grave disabilità Ereditarie: - Distrofie muscolari - Miopatie congenite - Miopatie mitocondriali
MIOPATIE Malattie del muscolo scheletrico da primitiva compromissione strutturale e/o funzionale. Spesso causa di grave disabilità Ereditarie: - Distrofie muscolari - Miopatie congenite - Miopatie mitocondriali
DISTROFIA FACIO SCAPOLO-OMERALE (FSH
 DISTROFIA FACIO SCAPOLO-OMERALE (FSH = facioscapulohomeral muscolar daystrophy) Detta anche distrofia di Landouzy-Dejerine che per primi la descrissero nel 1884. Definizione La distrofia FSH è la terza
DISTROFIA FACIO SCAPOLO-OMERALE (FSH = facioscapulohomeral muscolar daystrophy) Detta anche distrofia di Landouzy-Dejerine che per primi la descrissero nel 1884. Definizione La distrofia FSH è la terza
Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
 Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked") Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Trasmissione ereditaria di un singolo gene (eredità monofattoriale) Mendeliana Autosomica Dominante (AD) Autosomica Recessiva (AR) X-linked Recessiva (X-linked R) X-linked Dominante (X-linked D) Y-linked
Lettere di una madre al proprio figlio
 A te Lettere di una madre al proprio figlio Ilaria Baldi A TE Lettere di una madre al proprio figlio Diario www.booksprintedizioni.it Copyright 2015 Ilaria Baldi Tutti i diritti riservati All amore più
A te Lettere di una madre al proprio figlio Ilaria Baldi A TE Lettere di una madre al proprio figlio Diario www.booksprintedizioni.it Copyright 2015 Ilaria Baldi Tutti i diritti riservati All amore più
Patologie da mutazioni dinamiche
 Patologie da mutazioni dinamiche Mutazioni dinamiche mutazioni progressive, nei tessuti e nelle generazioni, di ripetizioni instabili Alleli normali Pre-mutazioni Mutazioni Le alterazioni fenotipiche (patologia)
Patologie da mutazioni dinamiche Mutazioni dinamiche mutazioni progressive, nei tessuti e nelle generazioni, di ripetizioni instabili Alleli normali Pre-mutazioni Mutazioni Le alterazioni fenotipiche (patologia)
L integrazione scolastica degli allievi con Distrofia Muscolare Duchenne o Becker
 L integrazione scolastica degli allievi con Distrofia Muscolare Duchenne o Becker Raffaela Maggi USP - Ancona Falconara Marittima 16.12.08 Sotto il termine DISTROFIA MUSCOLARE si raccolgono un gruppo di
L integrazione scolastica degli allievi con Distrofia Muscolare Duchenne o Becker Raffaela Maggi USP - Ancona Falconara Marittima 16.12.08 Sotto il termine DISTROFIA MUSCOLARE si raccolgono un gruppo di
Frequenza: E' la più frequente tra le distrofie muscolari, colpisce circa 1 su 3500 maschi nati.
 DISTROFIA MUSCOLARE DI DUCHENNE Frequenza: E' la più frequente tra le distrofie muscolari, colpisce circa 1 su 3500 maschi nati. Definizione Con distrofia si intende una malattia genetica che causa una
DISTROFIA MUSCOLARE DI DUCHENNE Frequenza: E' la più frequente tra le distrofie muscolari, colpisce circa 1 su 3500 maschi nati. Definizione Con distrofia si intende una malattia genetica che causa una
CONTRIBUTO DELLA FISIOTERAPIA ALLA GESTIONE DEL BAMBINO CON MALATTIA NEUROMUSCOLARE
 CONTRIBUTO DELLA FISIOTERAPIA ALLA GESTIONE DEL BAMBINO CON MALATTIA NEUROMUSCOLARE Dispensa realizzata in occasione del CORSO DI FORMAZIONE PER CAREGIVER che assistono minori con patologie neuromuscolari
CONTRIBUTO DELLA FISIOTERAPIA ALLA GESTIONE DEL BAMBINO CON MALATTIA NEUROMUSCOLARE Dispensa realizzata in occasione del CORSO DI FORMAZIONE PER CAREGIVER che assistono minori con patologie neuromuscolari
5 modulo didattico - Patologia cromosomica.
 5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
5 modulo didattico - Patologia cromosomica. G0 IL CICLO CELLULARE DI UNA CELLULA DI MAMMIFERO Avviene ogni volta che la cellula si divide Le tappe fondamentali del processo sono: Separazione dei due filamenti
Presentazione progetti di ricerca Struttura Complessa Neuropsichiatria Infantile
 Presentazione progetti di ricerca Struttura Complessa Neuropsichiatria Infantile Studio Epidemiologico su Mucopolisaccaridosi Ricerca Geni Coinvolti in alcune forme di paraplegia spastica ereditaria Screening
Presentazione progetti di ricerca Struttura Complessa Neuropsichiatria Infantile Studio Epidemiologico su Mucopolisaccaridosi Ricerca Geni Coinvolti in alcune forme di paraplegia spastica ereditaria Screening
PRODA Istituto di Diagnostica Clinica
 Caratterizzazione molecolare delle Distrofie Muscolari di Duchenne (DMD) e di Becker (BMD): diagnosi di malattia e studi familari L Proda esegue le indagini molecolari per le Distrofie muscolari di Duchenne
Caratterizzazione molecolare delle Distrofie Muscolari di Duchenne (DMD) e di Becker (BMD): diagnosi di malattia e studi familari L Proda esegue le indagini molecolari per le Distrofie muscolari di Duchenne
scaricato da www.sunhope.it
 Distrofie muscolari Malattie geneticamente determinate ed a decorso cronico progressivo, riconducibili dipendenti da un processo degenerativo primario che interessa il muscolo scheletrico. Classificazione
Distrofie muscolari Malattie geneticamente determinate ed a decorso cronico progressivo, riconducibili dipendenti da un processo degenerativo primario che interessa il muscolo scheletrico. Classificazione
ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136)
 (OMIM #607136)") ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136) Il gene implicato nella SCA17 è il gene TATA box-binding protein (TBP) che fa parte del complesso della RNA polimerasi II ed è essenziale per dare inizio
ATASSIA SPINOCEREBELLARE 17 (SCA17) (OMIM #607136) Il gene implicato nella SCA17 è il gene TATA box-binding protein (TBP) che fa parte del complesso della RNA polimerasi II ed è essenziale per dare inizio
Le distrofie muscolari
 Le distrofie muscolari Maurizio Moggio UOD Malattie Neuromuscolari e Rare Fondazione IRCCS Ca Granda Ospedale Maggiore Policlinico Milano 34 Congresso Nazionale di Antibioticoterapia in età pediatrica
Le distrofie muscolari Maurizio Moggio UOD Malattie Neuromuscolari e Rare Fondazione IRCCS Ca Granda Ospedale Maggiore Policlinico Milano 34 Congresso Nazionale di Antibioticoterapia in età pediatrica
La prevenzione delle Malattie Rare e la genetica
 Centro di Riferimento Regionale per le Malattie Rare II Clinica Pediatrica Ospedale Regionale per le Microcitemie La prevenzione delle Malattie Rare e la genetica Dott.ssa Francesca Meloni Dott.ssa Paola
Centro di Riferimento Regionale per le Malattie Rare II Clinica Pediatrica Ospedale Regionale per le Microcitemie La prevenzione delle Malattie Rare e la genetica Dott.ssa Francesca Meloni Dott.ssa Paola
Scala fenotipica. Dominante o recessivo? fenotipo. fenotipo. fenotipo A 1 A 1 A 2 A 2 A 1 A 2
 fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
fenotipo Dominante o recessivo? Scala fenotipica fenotipo A 1 A 1 A 2 A 2 fenotipo A 1 A 2 A 1 dominante A 1 e A 2. codominanti A 2 dominante A 1 dominanza incompleta A 2 dominanza incompleta Nei casi
Piattaforma per studi su larga scala di eventi molecolari associati a patologie umane: applicazioni per la scoperta di nuovi farmaci
 Piattaforma per studi su larga scala di eventi molecolari associati a patologie umane: applicazioni per la scoperta di nuovi farmaci Roberto Ravazzolo Direttore Nicoletta Pedemonte Dirigente Biologo U.O.C.
Piattaforma per studi su larga scala di eventi molecolari associati a patologie umane: applicazioni per la scoperta di nuovi farmaci Roberto Ravazzolo Direttore Nicoletta Pedemonte Dirigente Biologo U.O.C.
Aggiornamenti in ambito genetico
 10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
10 Congresso Nazionale sulla Sindrome di Cornelia de Lange 1-4 novembre 2012 - Gabicce Aggiornamenti in ambito genetico Cristina Gervasini Genetica Medica Dip. Scienze della Salute Università degli Studi
A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo
 2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
2 Convegno Nazionale Sindrome di Rubinstein Taybi Lodi, 17 19 maggio 2013 A cosa serve al clinico e alla famiglia conoscere il difetto di base? Correlazione genotipo fenotipo Donatella Milani Cristina
Unità Operativa di GENETICA MEDICA. Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara. Prestazioni richieste
 Unità Operativa di GENETICA MEDICA Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara Direttore: Prof.ssa Alessandra Ferlini www.ospfe.it/geneticamedica LABORATORIO DI GENETICA MOLECOLARE Responsabile:
Unità Operativa di GENETICA MEDICA Azienda Ospedaliera Universitaria S.Anna AOSPFE, Ferrara Direttore: Prof.ssa Alessandra Ferlini www.ospfe.it/geneticamedica LABORATORIO DI GENETICA MOLECOLARE Responsabile:
Malattie neuromuscolari: definizione
 Malattie muscolari Malattie neuromuscolari: definizione Si definiscono malattie muscolari quelle condizioni patologiche caratterizzate da sintomi e segni attribuibili ad alterazioni biochimiche, elettrofisiologiche
Malattie muscolari Malattie neuromuscolari: definizione Si definiscono malattie muscolari quelle condizioni patologiche caratterizzate da sintomi e segni attribuibili ad alterazioni biochimiche, elettrofisiologiche
Capitolo 5_d URGENZE MEDICHE ADULTO
 Capitolo 5_d URGENZE MEDICHE ADULTO 1 OBIETTIVI RICONOSCERE LE PRINCIPALI PATOLOGIE NEUROLOGICHE CHE PROVOCANO ALTERAZIONI DELLA COSCIENZA IDENTIFICARE I SEGNI E SINTOMI DI: SINCOPE, CONVULSIONI, MENINGITE,
Capitolo 5_d URGENZE MEDICHE ADULTO 1 OBIETTIVI RICONOSCERE LE PRINCIPALI PATOLOGIE NEUROLOGICHE CHE PROVOCANO ALTERAZIONI DELLA COSCIENZA IDENTIFICARE I SEGNI E SINTOMI DI: SINCOPE, CONVULSIONI, MENINGITE,
4 modulo didattico - Modalità di trasmissione delle malattie
 4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
4 modulo didattico - Modalità di trasmissione delle malattie monogeniche. L analisi dell albero genealogico: uno strumento indispensabile della genetica medica I SIMBOLI DELL ALBERO GENEALOGICO L ANEMIA
Università degli studi di Cagliari
 Università degli studi di Cagliari DIPARTIMENTO DI SCIENZE BIOMEDICHE E BIOTECNOLOGICHE DOTTORATO DI RICERCA TERAPIA PEDIATRICA E FARMACOLOGIA DELLO SVILUPPO XIX CICLO DISCIPLINA: SCIENZE MEDICHE PEDIATRIA
Università degli studi di Cagliari DIPARTIMENTO DI SCIENZE BIOMEDICHE E BIOTECNOLOGICHE DOTTORATO DI RICERCA TERAPIA PEDIATRICA E FARMACOLOGIA DELLO SVILUPPO XIX CICLO DISCIPLINA: SCIENZE MEDICHE PEDIATRIA
Sindrome di Down. Aspetti genetici, fisici, motori e medici. www.fluitekruidje.com
 Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Sindrome di Down Aspetti genetici, fisici, motori e medici www.diagnosiprenatale.it www.lucinafoundation.org www.fluitekruidje.com Corso di Disabilità cognitive Prof. Renzo Vianello - Università di Padova
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide
 Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
Aneuploidie: il numero dei cromosomi non è un multiplo esatto del normale assetto aploide Aneuploidie Nullisomia: 2n-2 (morte preimpianto) Monosomia: : 2n-1 (generalmente morte embrionale) Trisomia: :
Sclerosi Laterale Amiotrofica (SLA) www.fisiokinesiterapia.biz
 www.fisiokinesiterapia.biz") Sclerosi Laterale Amiotrofica (SLA) www.fisiokinesiterapia.biz Sclerosi Laterale Amiotrofica (SLA) La più grave fra le malattie che colpiscono il motoneurone: sclerosi atrofia gliotica laterale cordoni
Sclerosi Laterale Amiotrofica (SLA) www.fisiokinesiterapia.biz Sclerosi Laterale Amiotrofica (SLA) La più grave fra le malattie che colpiscono il motoneurone: sclerosi atrofia gliotica laterale cordoni
PRODA Istituto di Diagnostica Clinica
 PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
PRODA Istituto di Diagnostica Clinica Sezione di Citogenetica e Genetica molecolare Responsabile: Dott. Guglielmo Sabbadini Specialista in Genetica Medica Informazioni per la diagnosi molecolare di sordita
SCREENING NEONATALE RISPARMIO PER IL SERVIZIO SANITARIO NAZIONALE?
 SCREENING NEONATALE GENETICO RISPARMIO PER IL SERVIZIO SANITARIO NAZIONALE? Pietro Chiurazzi Istituto di Genetica Medica Facoltà di Medicina e Chirurgia Università Cattolica Associazione Culturale Giuseppe
SCREENING NEONATALE GENETICO RISPARMIO PER IL SERVIZIO SANITARIO NAZIONALE? Pietro Chiurazzi Istituto di Genetica Medica Facoltà di Medicina e Chirurgia Università Cattolica Associazione Culturale Giuseppe
Anomalie cromosomiche
 Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
Anomalie cromosomiche costituzionali acquisite nelle cellule germinali nelle cellule somatiche Le alterazioni che avvengono nelle cellule germinali se compatibili con la vita porteranno alla nascita di
Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna
 Eredità mitocondriale Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna I mitocondri dello spermatozoo non entrano a far
Eredità mitocondriale Si tratta di una eredità non mendeliana con una trasmisione matrilineare I caratteri si trasmettono principalmente per via materna I mitocondri dello spermatozoo non entrano a far
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali
 CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
CATCH 22 e altre indagini genetiche nel counseling dopo diagnosi di malformazioni fetali Milano Marittima 05/06/2010 Dr. Francesco Pigliapoco Lab. Citogenetica A.O.U.-Salesi- Ancona Le sindromi DiGeorge
La Malattia di Alzheimer. Manuel Soldato
 La Malattia di Alzheimer Manuel Soldato Caratteristiche Più frequente forma di demenza Deterioramento ingravescente delle capacità cognitive Comparsa di disturbi comportamentali e dell affettività Progressiva
La Malattia di Alzheimer Manuel Soldato Caratteristiche Più frequente forma di demenza Deterioramento ingravescente delle capacità cognitive Comparsa di disturbi comportamentali e dell affettività Progressiva
DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE
 DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE Istituto di Genetica UNIVERSITA DEGLI STUDI DI UDINE Address: Istituto di Genetica DSTB - Università degli Studi di Udine P.le Kolbe,1-33100 Udine - ITALIA
DIPARTIMENTO DI SCIENZE E TECNOLOGIE BIOMEDICHE Istituto di Genetica UNIVERSITA DEGLI STUDI DI UDINE Address: Istituto di Genetica DSTB - Università degli Studi di Udine P.le Kolbe,1-33100 Udine - ITALIA
Bari, 27 Febbraio 2010 Nicoletta Resta Dipartimento di Biomedicina dell Età Evolutiva UOC Lab. Genetica Medica INDAGINI GENETICHE: QUANDO E PERCHE
 Bari, 27 Febbraio 2010 Nicoletta Resta Dipartimento di Biomedicina dell Età Evolutiva UOC Lab. Genetica Medica INDAGINI GENETICHE: QUANDO E PERCHE EREDITA MENDELIANA CLASSICA ~ 600 E C M 10.000 Enzimi
Bari, 27 Febbraio 2010 Nicoletta Resta Dipartimento di Biomedicina dell Età Evolutiva UOC Lab. Genetica Medica INDAGINI GENETICHE: QUANDO E PERCHE EREDITA MENDELIANA CLASSICA ~ 600 E C M 10.000 Enzimi
Il vostro bambino e lo Screening Neonatale
 Il vostro bambino e lo Screening Neonatale Guida per i Genitori A cura di: Centro Fibrosi Cistica e Centro Malattie Metaboliche AOU A. Meyer, Firenze Cari genitori, la Regione Toscana, secondo un programma
Il vostro bambino e lo Screening Neonatale Guida per i Genitori A cura di: Centro Fibrosi Cistica e Centro Malattie Metaboliche AOU A. Meyer, Firenze Cari genitori, la Regione Toscana, secondo un programma
Le lesioni muscolari
 Le lesioni muscolari Cenni di anatomia e fisiologia Una classificazione è utile solo se fornisce informazioni sulla natura della lesione, sul suo trattamento e sulla sua prognosi M. E. Muller TIPI DI MUSCOLI
Le lesioni muscolari Cenni di anatomia e fisiologia Una classificazione è utile solo se fornisce informazioni sulla natura della lesione, sul suo trattamento e sulla sua prognosi M. E. Muller TIPI DI MUSCOLI
La genetica della Sindrome di Prader-Willi
 La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
La genetica della Sindrome di Prader-Willi Dott.ssa Milan Gabriella Laboratorio Endocrino Metabolico DIPARTIMENTO DI SCIENZE MEDICHE E CHIRURGICHE Università degli Studi di Padova Clinica Medica 3 Prader,
Screening dei soggetti a rischio e diagnostica molecolare
 Screening dei soggetti a rischio e diagnostica molecolare Daniela Furlan U.O. Anatomia Patologica Varese, 3 luglio 2012 Il carcinoma pancreatico Patogenesi Suscettibilità genetica Implicazioni diagnostiche
Screening dei soggetti a rischio e diagnostica molecolare Daniela Furlan U.O. Anatomia Patologica Varese, 3 luglio 2012 Il carcinoma pancreatico Patogenesi Suscettibilità genetica Implicazioni diagnostiche
Lo sviluppo del cancro è un processo complesso che coinvolge parecchi cambiamenti nella stessa cellula staminale. Poiché tutte le cellule staminali
 Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
Tumore Cos è il tumore? Il tumore o neoplasia (dal greco neo,, nuovo, e plasìa,, formazione), o cancro se è maligno, è una classe di malattie caratterizzate da una incontrollata riproduzione di alcune
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico. Volume sulle Disabilità intellettive, cap. 5.
 Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
Renzo Vianello Disturbi Pervasivi dello Sviluppo o Spettro autistico Volume sulle Disabilità intellettive, cap. 5. I disturbi pervasivi dello sviluppo si caratterizzano per la presenza di disabilità almeno
SCAMBI tra due coppie di cromosomi NON omologhi
 TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
TRASLOCAZIONI RECIPROCHE SCAMBI tra due coppie di cromosomi NON omologhi La traslocazione tra i cromosomi X e 21 può interrompere la sequenza del gene DMD e causare la manifestazione della DISTROFIA MUSCOLARE
La Sclerosi Multipla (SM) è una patologia infiammatorio-degenerativa del Sistema Nervoso Centrale a decorso cronico che colpisce il giovane adulto.
 è una patologia infiammatorio-degenerativa del Sistema Nervoso Centrale a decorso cronico che colpisce il giovane adulto.") Sclerosi multipla FONTE : http://www.medicitalia.it/salute/sclerosi-multipla La Sclerosi Multipla (SM) è una patologia infiammatorio-degenerativa del Sistema Nervoso Centrale a decorso cronico che colpisce
Sclerosi multipla FONTE : http://www.medicitalia.it/salute/sclerosi-multipla La Sclerosi Multipla (SM) è una patologia infiammatorio-degenerativa del Sistema Nervoso Centrale a decorso cronico che colpisce
Epidemiologia delle ipoacusie infantili
 Epidemiologia delle ipoacusie infantili 78 milioni di persone nel mondo presentano una ipoacusia moderata (>40 db) nell orecchio migliore e 364 milioni di persone hanno una ipoacusia lieve (26-40 db).
Epidemiologia delle ipoacusie infantili 78 milioni di persone nel mondo presentano una ipoacusia moderata (>40 db) nell orecchio migliore e 364 milioni di persone hanno una ipoacusia lieve (26-40 db).
Sostegno e locomozione
 Sostegno e locomozione Uno scheletro è un apparato che deve svolgere almeno le seguenti funzioni: sostenere il corpo permettere il movimento proteggere organi interni Uno scheletro può essere: Idroscheletro
Sostegno e locomozione Uno scheletro è un apparato che deve svolgere almeno le seguenti funzioni: sostenere il corpo permettere il movimento proteggere organi interni Uno scheletro può essere: Idroscheletro
Prof. Pier Paolo Piccaluga Università di Bologna
 Prof. Pier Paolo Piccaluga Università di Bologna DNA: la molecola della vita L'acido desossiribonucleico (DNA) è un acido nucleico, presente nel nucleo delle cellule, che contiene le informazioni genetiche
Prof. Pier Paolo Piccaluga Università di Bologna DNA: la molecola della vita L'acido desossiribonucleico (DNA) è un acido nucleico, presente nel nucleo delle cellule, che contiene le informazioni genetiche
LA SINDROME DI VON HIPPEL-LINDAU. Conoscere per Curare
 LA SINDROME DI VON HIPPEL-LINDAU Conoscere per Curare 1) Che cos è VHL? La sindrome di Von Hippel-Lindau è una rara malattia a carattere ereditario che determina una predisposizione allo sviluppo di neoplasie
LA SINDROME DI VON HIPPEL-LINDAU Conoscere per Curare 1) Che cos è VHL? La sindrome di Von Hippel-Lindau è una rara malattia a carattere ereditario che determina una predisposizione allo sviluppo di neoplasie
GENETICA MENDELIANA NELL UOMO
 GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
GENETICA MENDELIANA NELL UOMO GENETICA FORMALE o GENETICA CLASSICA basata unicamente su risultati visibili di atti riproduttivi. È la parte più antica della genetica, risalendo agli esperimenti di Mendel
Le demenze. Manuel Soldato Geriatra RSA S. F. Cabrini - Codogno
 Le demenze Manuel Soldato Geriatra RSA S. F. Cabrini - Codogno LE DEMENZE DEFINIZIONE 1. La demenza definisce uno stato di progressivo decadimento delle funzioni cognitive prodotto da una patologia cerebrale
Le demenze Manuel Soldato Geriatra RSA S. F. Cabrini - Codogno LE DEMENZE DEFINIZIONE 1. La demenza definisce uno stato di progressivo decadimento delle funzioni cognitive prodotto da una patologia cerebrale
Regolazione dell attività cardiaca
 Il cuore Il cuore è l organo centrale del sistema cardiocircolatorio che con la sua contrazione regolare consente al sangue di fluire continuamente attraverso i vasi trasportando numerose sostanze in tutti
Il cuore Il cuore è l organo centrale del sistema cardiocircolatorio che con la sua contrazione regolare consente al sangue di fluire continuamente attraverso i vasi trasportando numerose sostanze in tutti
Determinazione del sesso Cromosomi sessuali
 Determinazione del sesso Cromosomi sessuali Negli Eucarioti un cromosoma del sesso è un cromosoma presente in forme diverse nei due sessi. Uno è un cromosoma "X", l'altro strutturalmente e funzionalmente
Determinazione del sesso Cromosomi sessuali Negli Eucarioti un cromosoma del sesso è un cromosoma presente in forme diverse nei due sessi. Uno è un cromosoma "X", l'altro strutturalmente e funzionalmente
Corso nursing Ortopedico. Assistenza infermieristica ai pazienti con artrite reumatoide. www.slidetube.it
 Corso nursing Ortopedico Assistenza infermieristica ai pazienti con artrite reumatoide DEFINIZIONE MALATTIA INFIAMMATORIA SU BASE IMMUNITARIA DEL TESSUTO CONNETTIVO,ESSENZIALMENTE POLIARTICOLARE AD EVOLUZIONE
Corso nursing Ortopedico Assistenza infermieristica ai pazienti con artrite reumatoide DEFINIZIONE MALATTIA INFIAMMATORIA SU BASE IMMUNITARIA DEL TESSUTO CONNETTIVO,ESSENZIALMENTE POLIARTICOLARE AD EVOLUZIONE
LA DIAGNOSI PRENATALE : PRESENTE E FUTURO
 LA DIAGNOSI PRENATALE : PRESENTE E FUTURO LA CONSULENZA GENETICA PRENATALE Fare Dr. Renato clic per Scarinci modificare lo stile del sottotitolo dello schema GENETICA CLINICA Costituisce la parte applicativa
LA DIAGNOSI PRENATALE : PRESENTE E FUTURO LA CONSULENZA GENETICA PRENATALE Fare Dr. Renato clic per Scarinci modificare lo stile del sottotitolo dello schema GENETICA CLINICA Costituisce la parte applicativa
Unità Operativa di GENETICA MEDICA. Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE. Responsabile Dott.ssa Alessandra Ferlini
 Unità Operativa di GENETICA MEDICA Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE Responsabile Dott.ssa Alessandra Ferlini Servizi forniti: Definizione genotipica e diagnosi molecolare
Unità Operativa di GENETICA MEDICA Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE Responsabile Dott.ssa Alessandra Ferlini Servizi forniti: Definizione genotipica e diagnosi molecolare
Gestione di pazienti con sindromi miopatiche Prof. Enrico Granieri Clinica Neurologica Università di Ferrara
 Gestione di pazienti con sindromi miopatiche Prof. Enrico Granieri Clinica Neurologica Università di Ferrara Coinvolgimento muscolo Fisioterapia Test obiettivo della forza scheletrico muscolare - ipotonia
Gestione di pazienti con sindromi miopatiche Prof. Enrico Granieri Clinica Neurologica Università di Ferrara Coinvolgimento muscolo Fisioterapia Test obiettivo della forza scheletrico muscolare - ipotonia
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali. Silvia Sansavini
 CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
CATCH 22 altre indagini genetiche nel counceling dopo diagnosi di malformazioni fetali Silvia Sansavini CATCH 22 FIBROSI CISTICA DISTROFIA MIOTONICA CATCH 22: microdelezione 22q11 Diagnosi Counceling Diagnosi
La trasmissione delle malattie genetiche. Anna Onofri
 La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
La trasmissione delle malattie genetiche Gli alberi genealogici Anna Onofri I simboli maggiormente utilizzati Le malattie genetiche Molte malattie genetiche sono legate ad un singolo gene e possono verificarsi
LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2
 LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2 PRIMA INFANZIA: LESIONI CUTANEE DALLO SVILUPPO CARATTERISTICO ( dermatite bollosa congenita )
LUCA ROCCHETTI U.O. GENETICA MEDICA AVR SINDROME DI BLOCH SULTZBERGER O INCONTINENTIA AI PIGMENTI DI TIPO 2 PRIMA INFANZIA: LESIONI CUTANEE DALLO SVILUPPO CARATTERISTICO ( dermatite bollosa congenita )
Le distrofie muscolari
 Le distrofie muscolari Lezione 10 Le distrofie muscolari Con questo termine vengono definite le miopatie di origine genetica la cui evoluzione è progressiva. Sul piano clinico la compromissione muscolare
Le distrofie muscolari Lezione 10 Le distrofie muscolari Con questo termine vengono definite le miopatie di origine genetica la cui evoluzione è progressiva. Sul piano clinico la compromissione muscolare
Il RUOLO DEL MEDICO DI MEDICINA GENERALE NELLA IDENTIFICAZIONE DELLE PERSONE CON SOSPETTA DEMENZA. T. Mandarino (MMG ASL RMA )
") Il RUOLO DEL MEDICO DI MEDICINA GENERALE NELLA IDENTIFICAZIONE DELLE PERSONE CON SOSPETTA DEMENZA T. Mandarino (MMG ASL RMA ) La Malattia di Alzheimer La malattia di Alzheimer è la forma più frequente
Il RUOLO DEL MEDICO DI MEDICINA GENERALE NELLA IDENTIFICAZIONE DELLE PERSONE CON SOSPETTA DEMENZA T. Mandarino (MMG ASL RMA ) La Malattia di Alzheimer La malattia di Alzheimer è la forma più frequente
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel
 Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
Il termine connettiviti indica un gruppo di malattie reumatiche, caratterizzate dall infiammazione cronica del tessuto connettivo, ossia di quel complesso tessuto con funzione di riempimento, sostegno
ll consumo di bevande alcoliche durante la gravidanza e l allattamento può avere effetti dannosi sulla salute del bambino.
 NON BERE BEVANDE ALCOLICHE IN GRAVIDANZA ED IN ALLATTAMENTO U.O. Consultorio Familiare Distretto n 1 e n 2 Ostetrica t CRISTINA FILIPPI Ostetrica BIANCA PIVA 1 Non bere bevande alcoliche in gravidanza
NON BERE BEVANDE ALCOLICHE IN GRAVIDANZA ED IN ALLATTAMENTO U.O. Consultorio Familiare Distretto n 1 e n 2 Ostetrica t CRISTINA FILIPPI Ostetrica BIANCA PIVA 1 Non bere bevande alcoliche in gravidanza
Fonti di cellule staminali pluripotenti: Le cellule staminali possiedono 2 caratteristiche principali: -La massa cellulare interna della blastocisti.
 possiedono 2 caratteristiche principali: Fonti di cellule staminali pluripotenti: -Si autorinnovano a lungo termine. -Danno origine a tutti i tipi di cellule differenziate. -La massa cellulare interna
possiedono 2 caratteristiche principali: Fonti di cellule staminali pluripotenti: -Si autorinnovano a lungo termine. -Danno origine a tutti i tipi di cellule differenziate. -La massa cellulare interna
La stadiazione della SLA: ingravescenza di malattia
 La stadiazione della SLA: ingravescenza di malattia Vincenzo Silani U.O.. Neurologia e Lab. Neuroscienze Università degli tudi di Milano IRCCS Istituto Auxologico Italiano - Milano Diagnosi di SLA: una
La stadiazione della SLA: ingravescenza di malattia Vincenzo Silani U.O.. Neurologia e Lab. Neuroscienze Università degli tudi di Milano IRCCS Istituto Auxologico Italiano - Milano Diagnosi di SLA: una
 I TRAUMI MUSCOLARI LE CONTUSIONI Contusioni più o meno gravi, almeno una volta nella vita, le abbiamo subite più o meno tutti. Esse, in parole povere, rappresentano il risultato di un evento traumatico
I TRAUMI MUSCOLARI LE CONTUSIONI Contusioni più o meno gravi, almeno una volta nella vita, le abbiamo subite più o meno tutti. Esse, in parole povere, rappresentano il risultato di un evento traumatico
Classificazione dei tessuti
 Tessuto nervoso Classificazione dei tessuti Tessuto nervoso Caratterizzato da eccitabilità e conduttività Costituito principalmente da due tipi di cellule: Cellule neuronali o neuroni che sono caratteristiche
Tessuto nervoso Classificazione dei tessuti Tessuto nervoso Caratterizzato da eccitabilità e conduttività Costituito principalmente da due tipi di cellule: Cellule neuronali o neuroni che sono caratteristiche
Che cos è la celiachia?
 Che cos è la celiachia? La celiachia è una malattia infiammatoria cronica dell intestino tenue, dovuta ad una intolleranza al glutine assunto attraverso la dieta. Il glutine è una proteina contenuta in
Che cos è la celiachia? La celiachia è una malattia infiammatoria cronica dell intestino tenue, dovuta ad una intolleranza al glutine assunto attraverso la dieta. Il glutine è una proteina contenuta in
1) FIBRE ROSSE A CONTRAZIONE LENTA (Tipo I) 2) FIBRE BIANCHE INTERMEDIE (Tipo IIa) 3) FIBRE BIANCHE A CONTRAZIONE RAPIDA (Tipo IIb)
 FIBRE ROSSE A CONTRAZIONE LENTA (Tipo I) 2) FIBRE BIANCHE INTERMEDIE (Tipo IIa) 3) FIBRE BIANCHE A CONTRAZIONE RAPIDA (Tipo IIb)") LE FIBRE MUSCOLARI La fibra muscolare è considerata l' unità funzionale del muscolo scheletrico o, più semplicemente, una delle tante cellule che lo compongono. Ogni muscolo è infatti formato da un certo
LE FIBRE MUSCOLARI La fibra muscolare è considerata l' unità funzionale del muscolo scheletrico o, più semplicemente, una delle tante cellule che lo compongono. Ogni muscolo è infatti formato da un certo
Ausili per la mobilità
 Ausili per la mobilità Le persone affette da SMA mostrano una debolezza generalizzata dei muscoli; tuttavia le modalità con le quali si manifesta la malattia sono differenti da individuo a individuo. Nelle
Ausili per la mobilità Le persone affette da SMA mostrano una debolezza generalizzata dei muscoli; tuttavia le modalità con le quali si manifesta la malattia sono differenti da individuo a individuo. Nelle
Insegnamento di Psicologia delle disabilità e dell integrazione scolastica (Prof.ssa Ottavia Albanese)
") Insegnamento di Psicologia delle disabilità e dell integrazione scolastica (Prof.ssa Ottavia Albanese) Benessere e compromissione delle capacità motorie: aspetti teorici e riflessioni Dott.ssa Nicoletta
Insegnamento di Psicologia delle disabilità e dell integrazione scolastica (Prof.ssa Ottavia Albanese) Benessere e compromissione delle capacità motorie: aspetti teorici e riflessioni Dott.ssa Nicoletta
Il Cancro è una malattia genetica
 Il Cancro è una malattia genetica PROCESSO MULTIFASICO Il Cancro è sempre genetico Talvolta il cancro è ereditario Ciò che viene ereditato non è la malattia, bensì la PREDISPOSIZIONE In assenza di ulteriori
Il Cancro è una malattia genetica PROCESSO MULTIFASICO Il Cancro è sempre genetico Talvolta il cancro è ereditario Ciò che viene ereditato non è la malattia, bensì la PREDISPOSIZIONE In assenza di ulteriori
NUOVE PROSPETTIVE PER IL RIPRISTINO DELLO SVILUPPO CEREBRALE NELLA SINDROME DI DOWN
 NUOVE PROSPETTIVE PER IL RIPRISTINO DELLO SVILUPPO CEREBRALE NELLA SINDROME DI DOWN GRUPPO DI RICERCA Coordinamento Università Partner Progetto realizzato con il contributo di Assicurazioni Generali ABSTRACT
NUOVE PROSPETTIVE PER IL RIPRISTINO DELLO SVILUPPO CEREBRALE NELLA SINDROME DI DOWN GRUPPO DI RICERCA Coordinamento Università Partner Progetto realizzato con il contributo di Assicurazioni Generali ABSTRACT
Espressione di geni specifici per un determinato tumore
 Espressione di geni specifici per un determinato tumore Paziente A: Non ha il cancro Espressione dei geni: Nessuna Biopsia Geni associati al cancro allo stomaco Paziente B: Ha un tumore allo stomaco Bassa
Espressione di geni specifici per un determinato tumore Paziente A: Non ha il cancro Espressione dei geni: Nessuna Biopsia Geni associati al cancro allo stomaco Paziente B: Ha un tumore allo stomaco Bassa
Progetto sulle esostosi multiple
 Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
Progetto sulle esostosi multiple PROGETTO SULLE ESOSTOSI MULTIPLE EREDITARIE Dott. Leonardo D Agruma Servizio di Genetica Medica - Dipartimento dell Età Evolutiva IRCCS Ospedale Casa Sollievo della Sofferenza
Il flusso dell informazione genetica. DNA -->RNA-->Proteine
 Il flusso dell informazione genetica DNA -->RNA-->Proteine Abbiamo visto i principali esperimenti che hanno dimostrato che il DNA è la molecola depositaria dell informazione genetica nella maggior parte
Il flusso dell informazione genetica DNA -->RNA-->Proteine Abbiamo visto i principali esperimenti che hanno dimostrato che il DNA è la molecola depositaria dell informazione genetica nella maggior parte
Dal DNA alle proteine: La trascrizione e la traduzione
 Dal DNA alle proteine: La trascrizione e la traduzione DNA RNA Trascrizione RNA PROTEINE Traduzione Dove avvengono? GLI EUCARIOTI I PROCARIOTI Cambell, Reece Biologia ZANICHELLI Trascrizione Sintesi di
Dal DNA alle proteine: La trascrizione e la traduzione DNA RNA Trascrizione RNA PROTEINE Traduzione Dove avvengono? GLI EUCARIOTI I PROCARIOTI Cambell, Reece Biologia ZANICHELLI Trascrizione Sintesi di
Le malattie recessive
 Le malattie recessive Frequente la perdita di funzione con aplosufficienza Nascono figli affetti in genere da genitori sani spesso consanguinei che sono eterozigoti ed hanno un rischio di avere un figlio
Le malattie recessive Frequente la perdita di funzione con aplosufficienza Nascono figli affetti in genere da genitori sani spesso consanguinei che sono eterozigoti ed hanno un rischio di avere un figlio
LA REAZIONE ALL'AMBIENTE: L'ATTIVITÀ MUSCOLARE TUTTI I MOVIMENTI NEL CORPO E DEL CORPO SONO LA CONSEGUENZA DI CONTRAZIONI E RILASCIAMENTI MUSCOLARI
 LA REAZIONE ALL'AMBIENTE: L'ATTIVITÀ MUSCOLARE TUTTI I MOVIMENTI NEL CORPO E DEL CORPO SONO LA CONSEGUENZA DI CONTRAZIONI E RILASCIAMENTI MUSCOLARI CLASSIFICAZIONI DEI MUSCOLI: STRIATO SCHELETRICO STRATO
LA REAZIONE ALL'AMBIENTE: L'ATTIVITÀ MUSCOLARE TUTTI I MOVIMENTI NEL CORPO E DEL CORPO SONO LA CONSEGUENZA DI CONTRAZIONI E RILASCIAMENTI MUSCOLARI CLASSIFICAZIONI DEI MUSCOLI: STRIATO SCHELETRICO STRATO
Relazione autonoma del paziente: questionario di registrazione medica (II) Il registro internazionale delle disferlinopatie
 Il registro internazionale delle disferlinopatie") Il registro internazionale delle disferlinopatie Questionario II Pagina 1 / 8 Relazione autonoma del paziente: questionario di registrazione medica (II) Il registro internazionale delle disferlinopatie
Il registro internazionale delle disferlinopatie Questionario II Pagina 1 / 8 Relazione autonoma del paziente: questionario di registrazione medica (II) Il registro internazionale delle disferlinopatie
Progetto Memento. Diagnosi precoce della malattia di Alzheimer e mantenimento della autonomia psicofisica
 Progetto Memento Diagnosi precoce della malattia di Alzheimer e mantenimento della autonomia psicofisica Progetto Memento Diagnosi precoce della malattia di Alzheimer e mantenimento della autonomia psicofisica
Progetto Memento Diagnosi precoce della malattia di Alzheimer e mantenimento della autonomia psicofisica Progetto Memento Diagnosi precoce della malattia di Alzheimer e mantenimento della autonomia psicofisica
Facioscapolomerale (FSHD) Emery-Dreifuss (EDMD) Duchenne (DMD) Becker (BMD) Cingoli (LGMD) Oculofaringea (OPMD)
 Emery-Dreifuss (EDMD) Duchenne (DMD) Becker (BMD) Cingoli (LGMD) Oculofaringea (OPMD)") Distrofie muscolari Sono un gruppo eterogeneo di malattie genetiche che interessano primariamente il muscolo scheletrico che progressivamente va incontro a processi degenerativi che ne alterano la struttura
Distrofie muscolari Sono un gruppo eterogeneo di malattie genetiche che interessano primariamente il muscolo scheletrico che progressivamente va incontro a processi degenerativi che ne alterano la struttura
Nel muscolo un incremento della concentrazione di Ca 2+ attiva la glicogenolisi.
 Nel muscolo un incremento della concentrazione di Ca 2+ attiva la glicogenolisi. Il Ca++ si lega alle subunità (calmodulina) della fosforilasi chinasi attivando l enzimal attiva inattiva attiva La fosforilasi
Nel muscolo un incremento della concentrazione di Ca 2+ attiva la glicogenolisi. Il Ca++ si lega alle subunità (calmodulina) della fosforilasi chinasi attivando l enzimal attiva inattiva attiva La fosforilasi
AMES CENTRO POLIDIAGNOSTICO STRUMENTALE SETTORE DI GENETICA CARDIOMIOPATIE
 CARDIOMIOPATIE Le cardiomiopatie costituiscono un gruppo eterogeneo di malattie del miocardio causate frequentemente da mutazioni genetiche. Tali patologie sono associate a disfunzioni elettriche e/o meccaniche
CARDIOMIOPATIE Le cardiomiopatie costituiscono un gruppo eterogeneo di malattie del miocardio causate frequentemente da mutazioni genetiche. Tali patologie sono associate a disfunzioni elettriche e/o meccaniche
IL MEDICO DI MEDICINA GENERALE E MEDICINA GENERALE L ALLARME EBOLA: CLINICA E MANAGEMENT DELLE EPIDEMIE DEL TERZO MILLENNIO NEL SETTING DELLA
 IL MEDICO DI MEDICINA GENERALE E L ALLARME EBOLA: CLINICA E MANAGEMENT DELLE EPIDEMIE DEL TERZO MILLENNIO NEL SETTING DELLA MEDICINA GENERALE CORSO DI FORMAZIONE A DISTANZA I CRITERI PER LA CLASSIFICAZIONE
IL MEDICO DI MEDICINA GENERALE E L ALLARME EBOLA: CLINICA E MANAGEMENT DELLE EPIDEMIE DEL TERZO MILLENNIO NEL SETTING DELLA MEDICINA GENERALE CORSO DI FORMAZIONE A DISTANZA I CRITERI PER LA CLASSIFICAZIONE
GENETICA seconda parte
 GENETICA seconda parte I cromosomi sono lunghe molecole di una sostanza l acido desossiribonucleico. DNA Il DNA è una lunga catena fatta da due lunghi fili avvolti su se stessi a doppia elica. Sembra una
GENETICA seconda parte I cromosomi sono lunghe molecole di una sostanza l acido desossiribonucleico. DNA Il DNA è una lunga catena fatta da due lunghi fili avvolti su se stessi a doppia elica. Sembra una
Psicopatologia dell anziano. Prof.ssa Elvira Schiavina 02 marzo 2016
 Psicopatologia dell anziano 02 marzo 2016 Invecchiamento e Psicopatogia Salute mentale dell anziano: nuovo campo della psicopatologia. Affinamento dei criteri di diagnosi, costruzione e validazione di
Psicopatologia dell anziano 02 marzo 2016 Invecchiamento e Psicopatogia Salute mentale dell anziano: nuovo campo della psicopatologia. Affinamento dei criteri di diagnosi, costruzione e validazione di
SISTEMI ENERGETICI. L ATP privato di uno dei suoi 3 radicali fosforici diventa ADP (adenosindifosfato).
.") SISTEMI ENERGETICI LE FONTI ENERGETICHE MUSCOLARI I movimenti sono resi possibili, dal punto di vista energetico, grazie alla trasformazione, da parte dei muscoli, dell energia chimica ( trasformazione
SISTEMI ENERGETICI LE FONTI ENERGETICHE MUSCOLARI I movimenti sono resi possibili, dal punto di vista energetico, grazie alla trasformazione, da parte dei muscoli, dell energia chimica ( trasformazione
I disturbi della mente
 a.a. 2005/2006 Laurea Specialistica in Fisica Corso di Fisica Medica 1 I disturbi della mente 16/5/2006 Evoluzione Degenerazione delle capacità cognitive Invecchiamento buono Invecchiamento fisiologico
a.a. 2005/2006 Laurea Specialistica in Fisica Corso di Fisica Medica 1 I disturbi della mente 16/5/2006 Evoluzione Degenerazione delle capacità cognitive Invecchiamento buono Invecchiamento fisiologico
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI
 DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
DIAGNOSI PRENATALE DEI DIFETTI CONGENITI DIFETTI CONGENITI Le anomalie congenite sono condizioni che si instaurano tra il momento del concepimento e la nascita. LA DIAGNOSI PRENATALE insieme di tecniche
GIORNATA CONTRO LA FIBRILLAZIONE ATRIALE. 3 ottobre 2015 Verona / Palermo
 CONTRO LA 3 ottobre 2015 Verona / Palermo 2015 POSTER - LOCANDINA 32015 OTTOBRE VERONA - P.zza Cittadella PALERMO - P.zza Bologni 10:00-18.00 32015 OTTOBRE VERONA - P.zza Cittadella PALERMO - P.zza Bologni
CONTRO LA 3 ottobre 2015 Verona / Palermo 2015 POSTER - LOCANDINA 32015 OTTOBRE VERONA - P.zza Cittadella PALERMO - P.zza Bologni 10:00-18.00 32015 OTTOBRE VERONA - P.zza Cittadella PALERMO - P.zza Bologni
ASPETTI MECCANICI DELLA CONTRAZIONE MUSCOLARE. FGE aa.2015-16
 ASPETTI MECCANICI DELLA CONTRAZIONE MUSCOLARE FGE aa.2015-16 ARGOMENTI INNERVAZIONE DEL MUSCOLO SCHELETRICO PLACCA NEURO MUSCOLARE UNITÀ NEURO MOTORIE TIPI DI CONTRAZIONE SCOSSE SEMPLICI RELAZIONI TENSIONE-LUNGHEZZA
ASPETTI MECCANICI DELLA CONTRAZIONE MUSCOLARE FGE aa.2015-16 ARGOMENTI INNERVAZIONE DEL MUSCOLO SCHELETRICO PLACCA NEURO MUSCOLARE UNITÀ NEURO MOTORIE TIPI DI CONTRAZIONE SCOSSE SEMPLICI RELAZIONI TENSIONE-LUNGHEZZA
Sindromi da dolore agli arti
 www.printo.it/pediatric-rheumatology/it/intro Sindromi da dolore agli arti Versione 2016 10. Osteocondrosi (sinonimi: osteonecrosi, necrosi avascolare) 10.1 Che cos è? La parola "osteocondrosi" significa
www.printo.it/pediatric-rheumatology/it/intro Sindromi da dolore agli arti Versione 2016 10. Osteocondrosi (sinonimi: osteonecrosi, necrosi avascolare) 10.1 Che cos è? La parola "osteocondrosi" significa
IL GLAUCOMA I A P B I T A L I A O N L U S P E R A M O R E D E L L A V I S T A
 IL GLAUCOMA I A P B I T A L I A O N L U S P E R A M O R E D E L L A V I S T A IL GLAUCOMA Che cos è? Campagna informativa per la prevenzione delle malattie oculari che possono compromettere la visione
IL GLAUCOMA I A P B I T A L I A O N L U S P E R A M O R E D E L L A V I S T A IL GLAUCOMA Che cos è? Campagna informativa per la prevenzione delle malattie oculari che possono compromettere la visione
EVOLUZIONE DEL LANCIO ETA -CORRELATA
 EVOLUZIONE DEL LANCIO ETA -CORRELATA Dott. Nicola de Gasperis Dott. Giovanni Di Giacomo Dott. Alberto Costantini Dott. Andrea De Vita Concordia Hospital for Special Surgery Rome - Italy Per gettare un
EVOLUZIONE DEL LANCIO ETA -CORRELATA Dott. Nicola de Gasperis Dott. Giovanni Di Giacomo Dott. Alberto Costantini Dott. Andrea De Vita Concordia Hospital for Special Surgery Rome - Italy Per gettare un
Il ruolo del Pediatra di Famiglia nei nati da madre diabetica. Dottor Roberto Cionini
 Il ruolo del Pediatra di Famiglia nei nati da madre diabetica Dottor Roberto Cionini A cosa deve fare attenzione il Pediatra di Famiglia in un bambino nato da madre diabetica. ANOMALIE NEL FETO INDOTTE
Il ruolo del Pediatra di Famiglia nei nati da madre diabetica Dottor Roberto Cionini A cosa deve fare attenzione il Pediatra di Famiglia in un bambino nato da madre diabetica. ANOMALIE NEL FETO INDOTTE
La Velocità. Terzo incontro P.O.M.S. Relatore Averoldi Michele. Venerdi 24 Ottobre 2014
 La Velocità Terzo incontro P.O.M.S. Relatore Averoldi Michele La velocità Definizione e funzionamento Velocità (1) La velocità è la capacità di compiere movimenti di una certa ampiezza in un tempo limitato.
La Velocità Terzo incontro P.O.M.S. Relatore Averoldi Michele La velocità Definizione e funzionamento Velocità (1) La velocità è la capacità di compiere movimenti di una certa ampiezza in un tempo limitato.
PAPILLOMA VIRUS UMANO E CARCINOMA DELLA CERVICE
 PAPILLOMA VIRUS UMANO E CARCINOMA DELLA CERVICE Lo screening come forma di prevenzione LE INFEZIONI DA HPV (PAPILLOMA VIRUS UMANO) Cos è l HPV? L HPV è una categoria di virus molto diffusa. La trasmissione
PAPILLOMA VIRUS UMANO E CARCINOMA DELLA CERVICE Lo screening come forma di prevenzione LE INFEZIONI DA HPV (PAPILLOMA VIRUS UMANO) Cos è l HPV? L HPV è una categoria di virus molto diffusa. La trasmissione
Valutazione delle condizioni mediche. Valutazione delle condizioni mediche
 Pagina 1 / 2 andare alla pagina: 1 6 Posizione del M/L Posizione del A/P Peso corporeo Determinare il peso corporeo. Prendere già in considerazione eventuali variazioni prevedibili (ad es. oscillazioni
Pagina 1 / 2 andare alla pagina: 1 6 Posizione del M/L Posizione del A/P Peso corporeo Determinare il peso corporeo. Prendere già in considerazione eventuali variazioni prevedibili (ad es. oscillazioni