Emoglobinopatie: screening e gestione clinica
|
|
|
- Valeria Landi
- 9 anni fa
- Просмотров:
Транскрипт
1 Emoglobinopatie: screening e gestione clinica (e cenni di consulenza genetica ) Comacchio, 1 Aprile 2016 Dott.ssa Stefania Bigoni Servizio di Genetica Medica, Azienda Ospedaliera Universitaria, Ferrara

2 Emoglobinopatie e talassemie gruppo di disordini ereditari dovuti rispettivamente a difetti qualitativi (emoglobinopatie) o quantitativi (talassemie) della sintesi dell emoglobina. Emery and Rimoin, 2007
 della sintesi dell emoglobina.")
3 PRINCIPALI CARATTERISTICHE DELL EMOGLOBINA A eme proteina tetramerica di p.m ~ daltons --->2 + 2 catene polipeptidiche catene α = 141 Aa catene β = 146 Aa eme eme eme Dove si trovano i geni per queste catene polipeptidiche?

4 gene

5 I geni per le catene globiniche sono situati sui cromosomi 11 e 16 I geni per le catene ε γ δ β sono linearmente situati sul cromosoma 11 I geni per le catene α sono situati sul cromosoma 16

6 La composizione in emoglobina dei lisati eritrocitari può essere quantificata mediante differenti metodiche (elettroforesi in gel di poliacrilamide, IEF, HPLC). Mediante tali metodiche è possibile identificare differenti emoglobine differenti tra loro per: struttura percentuali condizioni (epoca di sviluppo, patologie) in cui sono incrementate Hb A (α2β2) è il maggior componente dell emoglobina e generalmente può arrivare al 97% HbA1c (α2(β-n-glucosio)2) differisce dalla HbA dall aggiunta di un glucosio a livello dell NH2 terminale della catena beta. La percentuale di HbA1c è correlata alla concentrazione intracellulare di glucosio ed alla vita media dei globuli rossi. Valori normali fino a 5 %. Nei pazienti diabetici valore all incirca raddoppiato.

7 HbF (α2 γ2): - nel neonato 50-85% - dopo la nascita diminuisce rapidamente raggiungendo concentrazioni di circa il 10-15% a 4 mesi di vita e poi più lentamente verso i 3-4 anni di età arriva ai livelli dell adulto ~1%. Aumento: gravidanza, β talassemia, δβ talassemia, HPFH, drepanocitosi, tireotossicosi, anemia aplastica e megaloblastica, leucemia HbA2 (α2 2) circa il 2-3%: E eteregenea dovuta - aumenta in caso di beta talassemia, di anemia allamegaloblastica, presenza di duedi malattie autoimmuni, di problemi tiroidei, farmaci tipi di catene γ: Gγ e - diminuisce in caso di varianti emoglobinicheaγ (varianti delta globiniche), iposideremia e di anemia sideroblastica

8 ALTERAZIONI GENETICHE DELL EMOGLOBINA DISORDINI QUANTITATIVI (talassemia) -CATENE -CATENE DISORDINI QUALITATIVI (emoglobinopatie) SOLUBILITA STABILITA AFFINITA PER L O2
 SOLUBILITA STABILITA")
9 Difetti dell emoglobina possono causare differenti problematiche Compromissio ne qualitativa del sistema di trasporto dell ossigeno ai tessuti Hb ad alta affinità per l O2 Compromissi one quantitativa del sistema di trasporto dell ossigeno ai tessuti Induzione della tossicità sia a livello del GR che a livello dell' eritroblasta Talassemia Riduzione della performan ce circolatori a dei GR Drepanocito si

10 Talassemie Sono condizioni ad eziologia genetica caratterizzate da una minore od assente produzione di catene globiniche. Vengono classificate principalmente in: alfa talassemie beta talassemie delta talassemie

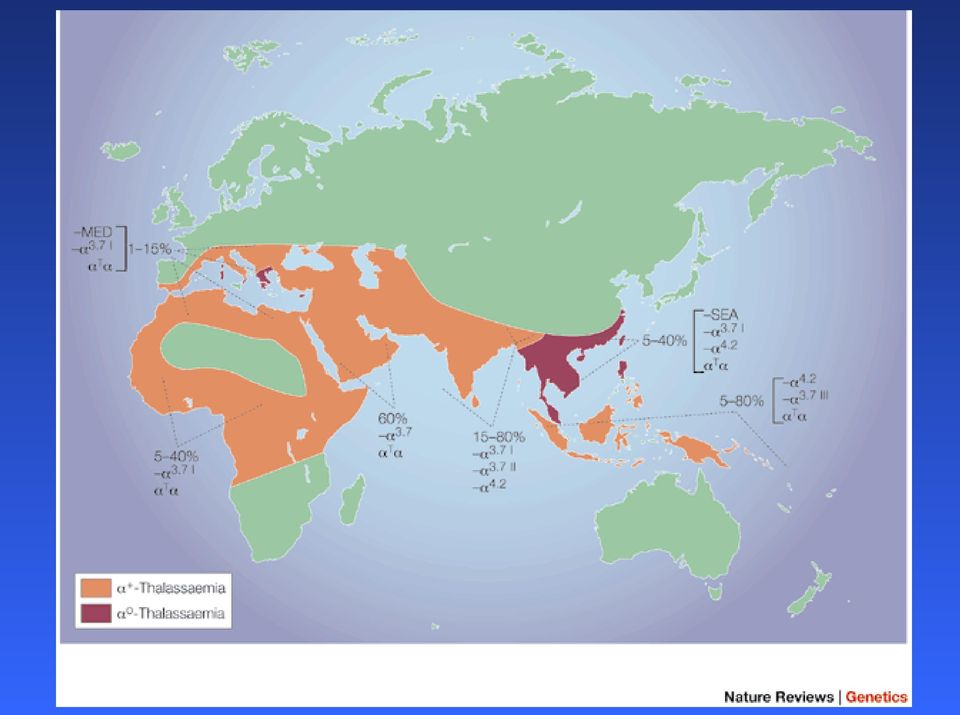
11 Talassemia causata principalmente da delezioni dei geni alfa: su ogni cromos sono presenti 2 copie del gene alfa : la delezione puo riguardare una o due copie, in cis o in trans Raggruppamento globine 16p13 Nel sud est asiatico sono presenti entrambi i tipi di delezione (un gene/due geni), mentre in Africa e praticamente assente l allele -- I genotipi possibili sono 6 e i fenotipi sono diversi a seconda delle combinazioni

12
13 Alfa talassemia Quando la produzione delle catene alfa è ridotta, l eccesso di catene beta e gamma può sua volta causare la formazione di tetrameri (Hb H and Hb Barts).

14 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

15 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

16 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

17 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

18 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

19 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale

20 Parametri ematologici Genotipo Hgb (g/dl) MCV (fl) RDW / Normale Normale Normale / Normale -/ - or - -/ / /


21 Modalità di trasmissione dell alfa talassemia
22
23
24 Talassemia Raggruppamento globine 11p15 G A Originata principalmente da mutazioni il cui effetto, attraverso me diversi, e quello di avere un deficit di catene. I siti di mutazione son caratteristici di una popolazione esone1introne1esone2 introne2 esone mutazioni nel promotore mutazioni puntiformi all interno degli esoni causano sostituzioni aa o fine mutazioni introni, alterazione splicing mutazione in siti di poliadenilazione
25 Talassemia talassemia: tutte le forme in cui manca la catena. l mrna puo mancare completamente, degradato o inattivo 0 talassemia: tutte le forme la catena e sottorappresentata l mrna puo non venire processato in maniera corretta o essere + poco rappresentato.
26

27 Le mutazioni della Talassemia nelle diverse popolazioni Sardegna: codone 39 da glutamina a stop proteina tronca 95 % codone 6 perdita di una base frameshift 2.1% codone 76 perdita di una base = 0.7% introne1 110 G A splicing alterato 0.5% introne2 745 C G == 0.4% Grecia: codone 39 introne1 1 introne1 6 introne1 110 introne2 745 da glutamina a stop proteina tronca 17.4 % G A splicing alterato 13.6 % T C splicing alterato 7.4 % G A == 43.7 % C G == 7.1%
28 La malattia si è diffusa quasi esclusivamente nelle zone in cui era presente la malaria, in quanto le caratteristiche anomale dei globuli rossi degli individui affetti da microcitemia impediscono o ostacolano il completamento del ciclo di vita del plasmodium, l'agente infettivo della malaria. In seguito i flussi migratori hanno portato la microcitemia a diffondersi anche in altre zone. In Italia le zone di maggior diffusione sono il Delta del Po, la Sardegna e la Sicilia con un'incidenza tra il 9 e il 14% della popolazione, per un totale di circa 7000 pazienti, ma si sta diffondendo anche in altre regioni quali Calabria, Puglia, Lombardia, Liguria e Piemonte.
29 -talassemia major X X X -talassemia intermedia X X X -talassemia minor tratto -talassemico -talassemia eterozigote X
30
31 Modalità di trasmissione della beta talassemia
32 GLOBIN-DISORDERS QUANTITATIVE DISORDERS (thalassemia) -CHAIN -CHAIN QUALITATIVE DISORDERS (structural hemoglobinopathie s) SOLUBILITY STABILITY Oxygen AFFINITY
33 DREPANOCITOSI (HBS) L emoglobinopatia più nota che compromette la solubilità dell Hb è la Drepanocitosi
34 Drepanocitosi: il gene Beta globin cluster region Gene Beta globinico Mutazione responsabile di HbS GAG>GTG CAC AAG Lysine HbC Gene Beta globinico Mutazione responsabile di HbC GAG>AAG

35 Drepanocitosi: il gene, la proteina The sickling balance POLYMERIZED HB SOLUBLE HB OXY-state, high PH, good hydration The sickling balance SOLUBLE HB POLYMERIZE D HB DEOXY-state, low PH, dehydration, fever
36 Drepanocitosi: il fenotipo o diversi fenotipi Principali Fenotipi: Omozigosi HbS Eterozigosi composta Sβ Eterozigosi composta Sβ+ Eterozigosi composta SC
37 Drepanocitosi: il fenotipo o diversi fenotipi OMOZIGOSI HBS Sintomatologia quadro clinico classico Fattori in grado in qualche modo di modulare il fenotipo A) Fattori correlati al β-globin cluster region (Aplotipi) B) Coesistenza di delezioni dei geni alfa globinici (delezioni che coinvolgono uno-due geni) C) Incremento dell HbF
38 Drepanocitosi: il fenotipo o diversi fenotipi T Eterozigosi composta Sβ (microdrepanocitosi) Eterozigosi composta βsβ Eterozigosi composta βsβ+ T T Fenotipo dipende da: Tipo di mutazione Beta talassemica β / β+ Aplotipo βs Nei β βs (vs βsβs): Persistenza della splenomegalia e crisi di sequestro splenico per lo più in adolescenza Ashley-Koch-a J Epidemiol, 2000
39 Drepanocitosi: il fenotipo o diversi fenotipi Eterozigosi composta SC Tendono ad avere un decorso più lieve Aumentato rischio di complicazioni tromboemboliche, retinopatia e necrosi renale papillare Ashley-Koch-a J Epidemiol, 2000
40 .LA CONSULENZA GENETICA NELLE TALASSEMIE ED EMOGLOBINOPATIE
41 .LA CONSULENZA GENETICA NELLE TALASSEMIE ED EMOGLOBINOPATIE POSTNATALE PRENATALE
42 Iter diagnostico: complesso coinvolgente diverse competenze da effettuarsi in tempi rapidi (epoca prenatale) Specialisti ginecologi Pediatri Equipe di consulenza genetica Laboratorio Analisi Laboratorio di Genetica Molecolare Laboratorio di Immunogenetica Servizio di Diagnosi Prenatale
43
44 Iter diagnostico: I step Consulenza genetica Raccolta di informazioni: Anamnesi personale e analisi dell albero familiare (altri familiari portatori o affetti, precedenti gravidanze esitate in idrope fetale) Area di provenienza (aree ad elevata frequenza di particolari difetti emoglobinici cfr. DeltaBeta tipo Sicilia) Accertamenti già eseguiti o da ripetersi in corso di consulenza Indici eritrocitari: G.R., Hb totale, MCV, MCH, RDW Parametri del ferro: ferritina, sideremia, % di saturazione della transferrina Assetto emoglobinico: % HbA2, % HbF, presenza di varianti Hb
45
46 Iter diagnostico: II step -> Diagnostica molecolare Metodiche utilizzate: Reverse Dot Blot Sequenza del gene Beta Ricerca mutazioni più frequenti a carico dei geni alfa globinici Sequenza dei geni alfa globinici MLPA (geni beta, alfa globinici) Identificazione del difetto emoglobinico dello stato di portatore in entrambi SI Stima quantitativa e qualitativa del rischio Proposta di Diagnosi prenatale NO Rivalutazione dei dati ematologici Considerazione altre ipotesi alternative
47 III step -> Diagnostica molecolare prenatale è la scelta prevalente delle coppie a rischio (fatta soprattutto nel 1 trimestre, ma anche in 2 trimestre con ricerca delle mutazioni parentali su DNA estratto da villi coriali o da amniociti) Previa somministrazione di specifico consenso informato con dettagliati i rischi di occorrenza della patologia, il quadro clinico atteso in base ai difetti molecolari (assicurato la disponibilità di eventuale colloquio con il pediatra del DH Talassemico) e i rischi di abortività connessi alla metodiche proposte Alle coppie con precedente figlio affetto offerta la possibilità di caratterizzazione HLA fetale in caso di feto sano o portatore
48 Caso-A Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 63 MCH 17 MCH 21,6 HbA2 4,9 HbA2 5,2 HbF HbF Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 0,9 Ipotesi: Portatore di trait beta talassemico 1,5 Analisi molecolare geni beta globinici: Eterozigosi Beta+IVS1-nt110 Counselling genetico: Rischio del 25% di feto affetto da T. Major SI Percorso di DP
49 Caso-B Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 85 MCH 17 MCH 29 HbA2 4,9 HbA2 3,9 HbF HbF Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 0,9 Ipotesi: Portatore di trait beta talassemico????? 1,3 Analisi molecolare geni beta globinici: Eterozigosi Beta+IVS1-nt101 Counselling genetico: Rischio del 25% di feto con fenotipo non dissimile dal portatore di trait beta talassemico NO. Percorso di DP
50 Caso-C Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 82 MCH 17 MCH 27 HbA2 4,9 HbA2 3,2 HbF HbF Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 0,9 Ipotesi: Portatore di????? 1,1 Analisi molecolare geni alfa globinici: Eterozigosi triplo alfa - anti 3.7 Counselling genetico: Rischio del 25% di feto Talassemia Intermedia
51
52 Caso-C Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 82 MCH 17 MCH 27 HbA2 4,9 HbA2 3,2 HbF HbF Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 0,9 Ipotesi: Portatore di????? 1,0 Analisi molecolare geni alfa globinici: Eterozigosi triplo alfa - anti 3.7 Counselling genetico: nella peggiore delle ipotesi.. Rischio del 25% di feto con Talassemia Intermedia?????????? Percorso di DP
53 III step -> Diagnostica molecolare prenatale Finalità. Rendere i singoli e le coppie consapevoli del rischio di patologia genetica per la prole, aiutandoli nelle loro decisioni riproduttive (counselling non direttivo) in altre parole: evitare la nascita di un bimbo affetto in coppie inconsapevoli del rischio e non evitare la nascita di un bambino affetto
54 Message to take home L approccio è per una patologia genetica autosomica-recessiva : ciò comporta un rischio solo se entrambi i partner hanno alterazioni dell Hb. Epoca preconcezionale: può essere conveniente fare sempre l esame completo ad almeno uno dei due partner, in caso di minimo dubbio approfondire l altro In gravidanza è meglio effettuare precocemente gli esami alla coppia Bisogna raccordare i tempi per completare la diagnosi per alterazioni dell Hb con i momenti utili per le procedure ostetriche connesse alla effettuazione dei. prelievi per D.P.; inizio: 11a, fine: 22a settimana gestazionale Importante la concordanza di tutti i dati in nostro possesso (dati familiari, accertamenti ematologici, cromatografia, molecolare) Attenzione errori di laboratorio!!!!!!!!
55 Grazie per l attenzione..
56
57 Caso-D Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 6,09 Hb Hb 11,6 11,6 MCV 62 MCV 90 MCH 22 MCH 30 HbA2 5,9 HbA2 1,4 HbF HbF 0,9 0,9 GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 63 MCH 17 MCH 21,6 HbA2 4,9 HbA2 2,9 HbF HbF 0,9 Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 Counselling genetico: Rischio del 25% di feto affetto da T. Major Ipotesi: Portatore di trait alfa talassemico 1,5 Analisi molecolare geni alfa globinici: negativa 1- Analisi molecolare geni beta globinici: Eterozigosi Beta+IVS1-nt MLPA cluster beta: eterozigosi per la delezione -7.2 Kb Corfu SI Percorso di DP
58 Caso-E Ipotesi: Portatore di trait beta talassemico GR 6,09 GR 5.00 Hb 10,6 Hb 11,8 MCV 57 MCV 75 MCH 17 MCH 25,6 HbA2 4,9 HbA2 2,7 HbF HbF Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 0,9 Ipotesi: Portatore di trait alfa talassemico 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Counselling genetico: Rischio del 25% di feto con doppia eterozigosi Beta cod 39 e delez. Alfa-3.7 NO Percorso di DP!!!!
59 Caso-F Ipotesi: Portatore di trait alfa talassemico GR 5.00 GR 5.00 Hb 11,8 Hb 11,8 MCV 75 MCV 75 MCH 25,6 MCH 25,6 HbA2 2,7 HbA2 2,7 HbF HbF 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Ipotesi: Portatore di trait alfa talassemico 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Counselling genetico: Rischio del 25% di feto con omozigosi per la del. alfa-3.7
60 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale Alcune forme di thalassemia sono dovute all instabilita dell mrn dell emoglobina: mutazione nel sito di poliadenilazione, perdita del c stop...
61 Caso-F Ipotesi: Portatore di trait alfa talassemico GR 5.00 GR 5.00 Hb 11,8 Hb 11,8 MCV 75 MCV 75 MCH 25,6 MCH 25,6 HbA2 2,7 HbA2 2,7 HbF HbF 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Ipotesi: Portatore di trait alfa talassemico 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Counselling genetico: Rischio del 25% di feto con omozigosi per la del. alfa-3.7 NO Percorso di DP!!
62 Caso-G Ipotesi: Portatore di trait alfa talassemico GR 6,39 GR 5,93 Hb Hb 13,6 MCV 66 13,6 MCV 67 MCH 20,3 MCH 21,9 HbA2 2,2 HbA2 2,7 HbF HbF Analisi molecolare geni alfa globinici: Eterozigosi delezione --FIL 0,4 Ipotesi: Portatore di trait alfa talassemico 1,4 Analisi molecolare geni alfa globinici: Eterozigosi delezione --FIL Counselling genetico: Rischio del 25% di feto con omozigosi per la del. --FIL
63 Genotipi e fenotipi nella atalassemia Assetto genomico N.Geni Stato eterozigoteeterozigote omozigote omozigote eterozigote 1 allele 1 allele nessun alleleomozigote wild-typewild type wild type nessun allele Anemia nessuna lieve wild-type! wild-type! lieve lieve grave HbH letale Alcune forme di thalassemia sono dovute all instabilita dell mrn dell emoglobina: mutazione nel sito di poliadenilazione, perdita del c stop...
64 Caso-G Ipotesi: Portatore di trait alfa talassemico GR 6,39 GR 5,93 Hb Hb MCV 13, ,6 MCV 67 MCH 20,3 MCH 21,9 HbA2 2,2 HbA2 2,7 HbF HbF Analisi molecolare geni alfa globinici: Eterozigosi delezione --FIL 0,4 Ipotesi: Portatore di trait alfa talassemico 1,4 Analisi molecolare geni alfa globinici: Eterozigosi delezione --FIL Counselling genetico: Rischio del 25% di feto con omozigosi per la del. --FIL SI Percorso di DP!!
65 HbH e HbBart disease
66 Caso-H Ipotesi: Portatore di trait alfa talassemico GR 6,39 GR 5.50 Hb 13,6 Hb 11,8 66 MCV 75 MCV MCH 20,3 MCH 25,6 HbA2 2,2 HbA2 2,7 HbF HbF Analisi molecolare geni alfa globinici: Eterozigosi delezione --FIL 0,4 Ipotesi: Portatore di trait alfa talassemico 1 Analisi molecolare geni alfa globinici: Eterozigosi del. alfa-3.7 Counselling genetico: Rischio del 25% di feto con eterozigosi composta per le del.-alfa 3.7/--FIL Forse. Percorso di DP
67 Caso-I Ipotesi: Portatore di HbS GR 6,39 GR 6,39 Hb Hb 13,6 13,6 MCV 82 MCV 83 MCH 27 MCH 28 HbA2 3,2 HbA2 3,3 HbF 0,4 HbF 0,4 HbS 40 HbS 38 Analisi molecolare geni beta globinici: Eterozigosi variante HbS Ipotesi: Portatore di HbS Analisi molecolare geni beta globinici: Eterozigosi variante HbS Counselling genetico:
68
69 Caso-H Ipotesi: Portatore di HbS GR 6,39 GR 6,39 Hb Hb 13,6 13,6 MCV 82 MCV 83 MCH 27 MCH 28 HbA2 3,2 HbA2 3,3 HbF 0,4 HbF 0,4 HbS 40 HbS 38 Analisi molecolare geni beta globinici: Eterozigosi variante HbS Counselling genetico: Rischio di feto affetto da Drepanocitosi: 25% Ipotesi: Portatore di HbS Analisi molecolare geni beta globinici: Eterozigosi variante HbS SI Percorso di DP!!
70 Caso-H Ipotesi: Portatore di trait beta talassemico GR 6,39 Hb 13,6 MCV 60 MCH 21 HbA2 5,2 HbF 2,4 Analisi molecolare geni beta globinici: Eterozigosi Beta cod 39 GR 6,39 Hb 13,6 MCV 83 MCH 28 Ipotesi: Portatore di HbS HbA2 3,3 HbF 0,4 HbS 38 Analisi molecolare geni beta globinici: Eterozigosi variante HbS Counselling genetico: Rischio del 25% di feto con eterozigosi composta per la mutazione Beta cod39 e la variante HbS: Microdrepanocitosi SI Percorso di DP!!
71 Message to take home L approccio è per una patologia genetica autosomica-recessiva : ciò comporta un rischio solo se entrambi i partner hanno alterazioni dell Hb. Epoca preconcezionale: può essere conveniente fare sempre l esame completo ad almeno uno dei due partner, in caso di minimo dubbio approfondire l altro In gravidanza è meglio effettuare precocemente gli esami alla coppia senza attendere la positività della gravida per l esame del partner Bisogna raccordare i tempi per completare la diagnosi per alterazioni dell Hb con i momenti utili per le procedure ostetriche connesse alla effettuazione dei. prelievi per D.P.; inizio: 11a, fine: 22a settimana gestazionale Importante la concordanza di tutti i dati in nostro possesso (dati familiari, accertamenti ematologici, cromatografia, molecolare) Attenzione errori di laboratorio!!!!!!!!
72 Grazie per l attenzione..
73
74 CONSULENZA GENETICA POSTNATALE Tipologie di consulenza principali 1- Consulenza genetica preconcezionale: coppie in cui entrambi i partner sono portatori di trait beta talassemico coppie in cui vi è il sospetto per uno/entrambi i partner di trait talassemico o di difetto emoglobinico coppie in cui uno dei due partner è affetto da T. Major (coppie PMA) 2- Consulenza genetica per inquadramento diagnostico: epoca infantile (soprattutto bambini adottati) nei quali agli accertamenti ematologici emerge il sospetto di anemia su base genetica
75 CONSULENZA GENETICA PRENATALE CHI sono i protagonisti? A- coppie in cui entrambi i partner sono portatori di trait beta talassemico Situazione più semplice!!!!!! Generalmente giungono precocemente in gravidanza Generalmente sono stati già caratterizzati molecolarmente in epoca prenatale Programmazione della diagnostica prenatale molecolare (spesso contestualmente alla diagnostica citogenetica)
76 CONSULENZA GENETICA PRENATALE CHI sono i protagonisti? B-coppie in cui vi è il sospetto per uno/entrambi i partner di trait talassemico o di emoglobinopatia INQUADRAMENTO DIAGNOSTICO E STIMA DEL RISCHIO RIPRODUTTIVO-PROGRAMMAZIONE DELLA DIAGNOSI PRENATALE Possibili complicazioni: Epoca tardiva di richiesta consulenza (!!!!!!!!!Importanza di fare informazione presso ginecologi del territorio) Parametri ematologici borderline per cui i difetti emoglobinici non sono facilmente sospettabili Coppie di extracomunitari: Epoca tardiva di richiesta consulenza Difetti emoglobinici non sovrapponibili a quelli del nostro territorio Cultura diversa: difficoltà nel far comprendere il quadro clinico della patologia e pertanto nel proporre una diagnostica prenatale specifica (ruolo dei mediatori culturali)
THALASSEMIA. Lezione 8. By NA
 THALASSEMIA Lezione 8 1 ancora eredita autosomica recessiva Perche le thalassemie? Perche sono un modello perfetto sia dal punto di vista della genetica formale che molecolare L emoglobina e stata una
THALASSEMIA Lezione 8 1 ancora eredita autosomica recessiva Perche le thalassemie? Perche sono un modello perfetto sia dal punto di vista della genetica formale che molecolare L emoglobina e stata una
Emoglobinopatie: il contributo del Laboratorio. Laura Michetti Laboratorio Analisi Clinico Cliniche AO Papa Giovanni XXIII LA Diagnostica Proteica
 Emoglobinopatie: il contributo del Laboratorio Laura Michetti Laboratorio Analisi Clinico Cliniche AO Papa Giovanni XXIII LA Diagnostica Proteica Scaletta degli argomenti Molecola emoglobinica: Emoglobinopatie
Emoglobinopatie: il contributo del Laboratorio Laura Michetti Laboratorio Analisi Clinico Cliniche AO Papa Giovanni XXIII LA Diagnostica Proteica Scaletta degli argomenti Molecola emoglobinica: Emoglobinopatie
Epidemiologia delle sindromi talassemiche ed emoglobinopatiche. Nel mondo e in casa nostra.
 Epidemiologia delle sindromi talassemiche ed emoglobinopatiche. Nel mondo e in casa nostra. Dott.ssa Maria Lerone 28/09/2013 Coop. Medici del Territorio Ara Nova Fiumicino . annually there are over 332000
Epidemiologia delle sindromi talassemiche ed emoglobinopatiche. Nel mondo e in casa nostra. Dott.ssa Maria Lerone 28/09/2013 Coop. Medici del Territorio Ara Nova Fiumicino . annually there are over 332000
Approccio Diagnostico delle Emoglobinopatie
 VII Congresso Nazionale S.I.T.E. IL FUTURO DELLE EMOGLOBINOPATIE Brindisi (Mesagne), 27-29 settembre 2012 Approccio Diagnostico delle Emoglobinopatie A. Giambona UOC di Ematologia per le Malattie Rare
VII Congresso Nazionale S.I.T.E. IL FUTURO DELLE EMOGLOBINOPATIE Brindisi (Mesagne), 27-29 settembre 2012 Approccio Diagnostico delle Emoglobinopatie A. Giambona UOC di Ematologia per le Malattie Rare
 Gruppo EME α β δ ϒ ζ ε A A2 α2β2 97-98 α2δ2 2-3 F Gower1 Gower2 Portland α2 ϒ2 < 1 ζ2ε2 0 α2ε2 0 ζ2ϒ2 0 Periodo fetale Periodo postnatale Varianti strutturali dell emoglobina Emoglobina anomala HbS
Gruppo EME α β δ ϒ ζ ε A A2 α2β2 97-98 α2δ2 2-3 F Gower1 Gower2 Portland α2 ϒ2 < 1 ζ2ε2 0 α2ε2 0 ζ2ϒ2 0 Periodo fetale Periodo postnatale Varianti strutturali dell emoglobina Emoglobina anomala HbS
Principali meccanismi attraverso i quali una mutazione provoca una patologia
 Principali meccanismi attraverso i quali una mutazione provoca una patologia v Perdita di funzione v Acquisizione di funzione v Produzione di un peptide che, oltre a non svolgere la normale funzione, interferisce
Principali meccanismi attraverso i quali una mutazione provoca una patologia v Perdita di funzione v Acquisizione di funzione v Produzione di un peptide che, oltre a non svolgere la normale funzione, interferisce
Giovanni Ivaldi Laboratorio di Genetica Umana Ospedali Galliera - Genova
 Giovanni Ivaldi Laboratorio di Genetica Umana Ospedali Galliera - Genova I cambiamenti della popolazione in Italia Costante crescita della popolazione straniera residente in Italia in milioni di soggetti
Giovanni Ivaldi Laboratorio di Genetica Umana Ospedali Galliera - Genova I cambiamenti della popolazione in Italia Costante crescita della popolazione straniera residente in Italia in milioni di soggetti
LA VALUTAZIONE DELL Hb F NEL PERCORSO DIAGNOSTICO DELLE EMOGLOBINOPATIE. Giannone Valentina Laboratorio di Genetica Medica
 LA VALUTAZIONE DELL Hb F NEL PERCORSO DIAGNOSTICO DELLE EMOGLOBINOPATIE Giannone Valentina Laboratorio di Genetica Medica Emoglobina Fetale Emoglobina Fetale Emoglobina Fetale Perché si misura l Hb F?
LA VALUTAZIONE DELL Hb F NEL PERCORSO DIAGNOSTICO DELLE EMOGLOBINOPATIE Giannone Valentina Laboratorio di Genetica Medica Emoglobina Fetale Emoglobina Fetale Emoglobina Fetale Perché si misura l Hb F?
Dr.ssa G. Barberio U.O. Medicina di Laboratorio Azienda Ulss 9 - Treviso. Brindisi 28/09/2012
 Dr.ssa G. Barberio U.O. Medicina di Laboratorio Azienda Ulss 9 - Treviso Brindisi 28/09/2012 Negli ultimi 15 anni, la diagnostica delle emoglobinopatie è diventata sempre più appannaggio dei Laboratori
Dr.ssa G. Barberio U.O. Medicina di Laboratorio Azienda Ulss 9 - Treviso Brindisi 28/09/2012 Negli ultimi 15 anni, la diagnostica delle emoglobinopatie è diventata sempre più appannaggio dei Laboratori
Il BPG è un importante modulatore dell affinità di Hb per l O 2. - curva iperbolica. - P50 = 1 mm Hg
 L Hb è una proteina allosterica che presenta: - regolazione omotropica O 2 è sia il normale ligando sia il modulatore omotropico - regolazione eterotropica H +, CO 2 e BPG sono modulatori eterotropici
L Hb è una proteina allosterica che presenta: - regolazione omotropica O 2 è sia il normale ligando sia il modulatore omotropico - regolazione eterotropica H +, CO 2 e BPG sono modulatori eterotropici
Le Emoglobinopatie e metodi diagnostici. Prof. Fabiana Passaro
 Le Emoglobinopatie e metodi diagnostici Prof. Fabiana Passaro Difetti di produzione di emoglobina su base ereditaria: - le TALASSEMIE, difetti quantitativi di produzione dell emoglobina a carico delle
Le Emoglobinopatie e metodi diagnostici Prof. Fabiana Passaro Difetti di produzione di emoglobina su base ereditaria: - le TALASSEMIE, difetti quantitativi di produzione dell emoglobina a carico delle
per la valutazione dell assetto emoglobinico Laboratorio Centralizzato Policlinico Universitario Ospedaliero S.Orsola Malpighi Bologna Rita Mancini
 1 anno esperienza UKNEQAS per la valutazione dell assetto emoglobinico Laboratorio Centralizzato Policlinico Universitario Ospedaliero S.Orsola Malpighi Bologna Rita Mancini Policlinico Ospedaliero Universitario:
1 anno esperienza UKNEQAS per la valutazione dell assetto emoglobinico Laboratorio Centralizzato Policlinico Universitario Ospedaliero S.Orsola Malpighi Bologna Rita Mancini Policlinico Ospedaliero Universitario:
Percorsi di laboratorio di II livello: raccomandazioni, criticità e consulenza genetica in diagnosi prenatale
 VII Congresso Nazionale S.I.T.E. IL FUTURO DELLE EMOGLOBINOPATIE Brindisi (Mesagne), 27-29 settembre 2012 Simposio Medicina di Laboratorio Percorsi di laboratorio di II livello: raccomandazioni, criticità
VII Congresso Nazionale S.I.T.E. IL FUTURO DELLE EMOGLOBINOPATIE Brindisi (Mesagne), 27-29 settembre 2012 Simposio Medicina di Laboratorio Percorsi di laboratorio di II livello: raccomandazioni, criticità
Talassemie ed Emoglobinopatie
 Talassemie ed Emoglobinopatie Corso di formazione professionale per Medici e Pediatri di famiglia Dott. Antonio Amato Centro Studi Microcitemie di Roma Fiumicino, 28 Settembre 2013 Ferrara, estate
Talassemie ed Emoglobinopatie Corso di formazione professionale per Medici e Pediatri di famiglia Dott. Antonio Amato Centro Studi Microcitemie di Roma Fiumicino, 28 Settembre 2013 Ferrara, estate
La diagnosi genetica della talassemia. Dott.ssa L.Pagano. Unità Operativa Microcitemia Dipartimento di Onco-Ematologia A.O.R.N. A.
 Diapositiva 1 La diagnosi genetica della talassemia Dott.ssa L.Pagano Unità Operativa Microcitemia Dipartimento di Onco-Ematologia A.O.R.N. A. Cardarelli Napoli 2 Dicembre 2005 Castel dell Ovo Sala Virgilio
Diapositiva 1 La diagnosi genetica della talassemia Dott.ssa L.Pagano Unità Operativa Microcitemia Dipartimento di Onco-Ematologia A.O.R.N. A. Cardarelli Napoli 2 Dicembre 2005 Castel dell Ovo Sala Virgilio
37 ANNI DI APPLICAZIONE DEL PIANO REGIONALE DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA
 37 ANNI DI APPLICAZIONE DEL PIANO REGIONALE DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA Anche quest'anno, a conclusione del precedente anno scolastico, nel mese di giugno, è
37 ANNI DI APPLICAZIONE DEL PIANO REGIONALE DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA Anche quest'anno, a conclusione del precedente anno scolastico, nel mese di giugno, è
TALASSEMIE (ANEMIA MEDITERRANEA)
") TALASSEMIE (ANEMIA MEDITERRANEA) Difetti genetici della sintesi di una o più catene globiniche normali Inadeguata produzione di emoglobina Anemia ipocromica-microcitica Sintesi bilanciata di catene globiniche,
TALASSEMIE (ANEMIA MEDITERRANEA) Difetti genetici della sintesi di una o più catene globiniche normali Inadeguata produzione di emoglobina Anemia ipocromica-microcitica Sintesi bilanciata di catene globiniche,
Il ruolo del medico del territorio negli screening per le emoglobinopatie: presentazione di casi clinici
 ASSOCIAZIONE NAZIONALE PER LA LOTTA CONTRO LE MICROCITEMIE IN ITALIA Dott.ssa Roberta Piscitelli TALASSEMIE ED EMOGLOBINOPATIE Corso di formazione professionale per Medici e Pediatri di famiglia Il ruolo
ASSOCIAZIONE NAZIONALE PER LA LOTTA CONTRO LE MICROCITEMIE IN ITALIA Dott.ssa Roberta Piscitelli TALASSEMIE ED EMOGLOBINOPATIE Corso di formazione professionale per Medici e Pediatri di famiglia Il ruolo
Screening delle emoglobinopatie
 Congresso Nazionale SIP - Bologna, 9 maggio 2013 Riunione del GLNBI Screening delle emoglobinopatie M. Zaffaroni, R. Rolla, M. Castagno, B. Grigollo, A. Boncompagni, C. Dellora, C. Bellomo, G. Bona Clinica
Congresso Nazionale SIP - Bologna, 9 maggio 2013 Riunione del GLNBI Screening delle emoglobinopatie M. Zaffaroni, R. Rolla, M. Castagno, B. Grigollo, A. Boncompagni, C. Dellora, C. Bellomo, G. Bona Clinica
UTILIZZO OTTIMALE DEI TEST DI II LIVELLO PER TALASSEMIA
 UTILIZZO OTTIMALE DEI TEST DI II LIVELLO PER TALASSEMIA Dr.ssa Anna Ravani Laboratorio di Genetica Molecolare Servizio di Genetica Medica Azienda Ospedaliera-Universitaria S.Anna FERRARA EMOGLOBINOPATIE:
UTILIZZO OTTIMALE DEI TEST DI II LIVELLO PER TALASSEMIA Dr.ssa Anna Ravani Laboratorio di Genetica Molecolare Servizio di Genetica Medica Azienda Ospedaliera-Universitaria S.Anna FERRARA EMOGLOBINOPATIE:
35 ANNI DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA
 35 ANNI DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA Si è concluso nel giugno 2010 il 35 screening dei portatori di microcitemia (l anomalia genetica responsabile dell anemia
35 ANNI DI PREVENZIONE DELL ANEMIA MEDITERRANEA NEL LAZIO RELAZIONE RIASSUNTIVA Si è concluso nel giugno 2010 il 35 screening dei portatori di microcitemia (l anomalia genetica responsabile dell anemia
Esperienze di un Centro di Riferimento per lo Screening delle Talassemie
 Esperienze di un Centro di Riferimento per lo Screening delle Talassemie 39 Congresso Nazionale SIBioC Nuovi orizzonti nella diagnostica delle talassemie e delle emoglobinopate Workshop Tosoh Rimini 2-5
Esperienze di un Centro di Riferimento per lo Screening delle Talassemie 39 Congresso Nazionale SIBioC Nuovi orizzonti nella diagnostica delle talassemie e delle emoglobinopate Workshop Tosoh Rimini 2-5
TALASSEMIA INTERMEDIA: casi clinici. Melania Serra Centro Microcitemie-Pediatria Ospedale San Luigi Gonzaga Orbassano (TO)
") TALASSEMIA INTERMEDIA: casi clinici Melania Serra Centro Microcitemie-Pediatria Ospedale San Luigi Gonzaga Orbassano (TO) Talassemia intermedia: definizione Anemia a esordio tardivo (sopra i 2 anni di
TALASSEMIA INTERMEDIA: casi clinici Melania Serra Centro Microcitemie-Pediatria Ospedale San Luigi Gonzaga Orbassano (TO) Talassemia intermedia: definizione Anemia a esordio tardivo (sopra i 2 anni di
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica
 Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Elementi di Patologia Generale Dott.ssa Samantha Messina Lezione: Patologia Genetica Anno accademico 2009/2010 I anno, II semestre CdL Infermieristica e Fisioterapia PATOLOGIA GENETICA Oggetto di studio
Principi dello screening raccomandati dalla World Health Organization
 EMOGLOBINE: DIAGNOSTICA, STANDARDIZZAZIONE, PROSPETTIVE Approcci diagnostici e problematiche in relazione ad uno screening neonatale Giovanni Ivaldi Laboratorio di Genetica - Settore Microcitemia Ospedali
EMOGLOBINE: DIAGNOSTICA, STANDARDIZZAZIONE, PROSPETTIVE Approcci diagnostici e problematiche in relazione ad uno screening neonatale Giovanni Ivaldi Laboratorio di Genetica - Settore Microcitemia Ospedali
Non è possibile che l analisi escluda in assoluto la probabilità di essere un portatore per le
 Nei pazienti affetti da FC il gene della CFTR è alterato, in genere a causa di mutazioni puntiformi. Queste alterazioni fanno sì che la proteina non venga più prodotta, o che non sia funzionale. Ciò provoca
Nei pazienti affetti da FC il gene della CFTR è alterato, in genere a causa di mutazioni puntiformi. Queste alterazioni fanno sì che la proteina non venga più prodotta, o che non sia funzionale. Ciò provoca
Patologie da analizzare
 Fasi cruciali Scelta della patologia da analizzare Scelta del campione da analizzare Scelta dell approccio da utilizzare Scelta della tecnica da utilizzare Analisi statistica del dati Conferme con approcci
Fasi cruciali Scelta della patologia da analizzare Scelta del campione da analizzare Scelta dell approccio da utilizzare Scelta della tecnica da utilizzare Analisi statistica del dati Conferme con approcci
Quando il fenotipo non è compatibile con il genotipo
 Quando il fenotipo non è compatibile con il genotipo Laura Perrone Dipartimento della Donna, del Bambino, di Chirurgia Generale e Specialistica Seconda Università degli Studi di Napoli Che cos è la Sindrome
Quando il fenotipo non è compatibile con il genotipo Laura Perrone Dipartimento della Donna, del Bambino, di Chirurgia Generale e Specialistica Seconda Università degli Studi di Napoli Che cos è la Sindrome
Mutazioni genetiche 2
 Mutazioni genetiche 2 Cosa sono le mutazioni? Le proteine sono in grado di svolgere la loro funzione solo se la loro sequenza amminoacidica è quella corretta. In caso contrario si possono generare delle
Mutazioni genetiche 2 Cosa sono le mutazioni? Le proteine sono in grado di svolgere la loro funzione solo se la loro sequenza amminoacidica è quella corretta. In caso contrario si possono generare delle
Thalassaemias. Thalassaemia Hb Variants. A group of blood deseases caused by a genetically determined. alteration in globin synthesis
 Approccio diagnostico delle Varianti Hb Giovanni Ivaldi Laboratorio di Genetica Umana - Settore Microcitemia Ospedali Galliera - Genova A group of blood deseases caused by a genetically determined reduction
Approccio diagnostico delle Varianti Hb Giovanni Ivaldi Laboratorio di Genetica Umana - Settore Microcitemia Ospedali Galliera - Genova A group of blood deseases caused by a genetically determined reduction
Le dimensioni ingannano
 Le dimensioni ingannano Natalia Scaramellini Centro Malattie Rare Università degli Studi di Milano Fondazione Cà Granda Ospedale Maggiore Policlinico Milano Cosa c è di nuovo per trattare l anemia Milano
Le dimensioni ingannano Natalia Scaramellini Centro Malattie Rare Università degli Studi di Milano Fondazione Cà Granda Ospedale Maggiore Policlinico Milano Cosa c è di nuovo per trattare l anemia Milano
 Anemie carenziali da fattori che regolano la sintesi del DNA Anemia con megaloblastosi midollare Iperbilirubinemia ed incremento della LDH per eritropoiesi inefficace Incremento sensibile dal volume corpuscolare
Anemie carenziali da fattori che regolano la sintesi del DNA Anemia con megaloblastosi midollare Iperbilirubinemia ed incremento della LDH per eritropoiesi inefficace Incremento sensibile dal volume corpuscolare
Le analisi prenatali
 L Amniocentesi L amniocentesi è l esame più utilizzato per la diagnosi prenatale di eventuali malattie genetiche. E una procedura che nella donna in gravidanza, tra la 15 e la 18 settimana, permette di
L Amniocentesi L amniocentesi è l esame più utilizzato per la diagnosi prenatale di eventuali malattie genetiche. E una procedura che nella donna in gravidanza, tra la 15 e la 18 settimana, permette di
FLOW CHART ANEMIE CRONICHE IN SARDEGNA
 1) Hb: diminuita = anemia FLOW CHART ANEMIE CRONICHE IN SARDEGNA Diminuito: anemia microcitica 2) MCV: Normale: anemia normocitica (acuta?) Aumentato: anemia macrocitica (carenza vit B?) chiediamo la ANEMIE
1) Hb: diminuita = anemia FLOW CHART ANEMIE CRONICHE IN SARDEGNA Diminuito: anemia microcitica 2) MCV: Normale: anemia normocitica (acuta?) Aumentato: anemia macrocitica (carenza vit B?) chiediamo la ANEMIE
Paola Selva Laboratorio Unico Metropolitano Azienda USL di Bologna
 Il nuovo Servizio UK NEQAS per la ricerca dell emoglobina su sangue neonatale Paola Selva Laboratorio Unico Metropolitano Azienda USL di Bologna Servizio UK NEQAS H EQA/PT per la ricerca dell emoglobina
Il nuovo Servizio UK NEQAS per la ricerca dell emoglobina su sangue neonatale Paola Selva Laboratorio Unico Metropolitano Azienda USL di Bologna Servizio UK NEQAS H EQA/PT per la ricerca dell emoglobina
Tappe essenziali nella proposta di diagnosi prenatale SAPERE PER SCEGLIERE
 DIAGNOSI PRENATALE la diagnosi prenatale è un insieme di indagini strumentali e di laboratorio finalizzate al monitoraggio dello stato di salute del concepito durante tutto il decorso della gravidanza
DIAGNOSI PRENATALE la diagnosi prenatale è un insieme di indagini strumentali e di laboratorio finalizzate al monitoraggio dello stato di salute del concepito durante tutto il decorso della gravidanza
sanguinamento (acuto/cronico) difetto intrinseco difetto estrinseco ANEMIE EMOLITICHE con GR normali cause estrinseche (acquisite)
 difetto intrinseco difetto estrinseco ANEMIE EMOLITICHE con GR normali cause estrinseche (acquisite)") sanguinamento (acuto/cronico) iporigenerativa progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico
sanguinamento (acuto/cronico) iporigenerativa progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico
1 modulo didattico - Impatto clinico delle malattie genetiche e
 1 modulo didattico - Impatto clinico delle malattie genetiche e fondamenti di genetica GENETICA MEDICA OBBIETTIVI FORMATIVI Conoscere le basi cellulari e molecolari dell eredità Conoscere le basi genetiche
1 modulo didattico - Impatto clinico delle malattie genetiche e fondamenti di genetica GENETICA MEDICA OBBIETTIVI FORMATIVI Conoscere le basi cellulari e molecolari dell eredità Conoscere le basi genetiche
Il cluster a-globinico: organizzazione, alterazioni molecolari e classificazione delle a-talassemie
 Il cluster a-globinico: organizzazione, alterazioni molecolari e classificazione delle a-talassemie Cristina Passarello U.O. Ematologia II Laboratorio per lo Studio e la Diagnosi Molecolare Prenatale di
Il cluster a-globinico: organizzazione, alterazioni molecolari e classificazione delle a-talassemie Cristina Passarello U.O. Ematologia II Laboratorio per lo Studio e la Diagnosi Molecolare Prenatale di
L informatizzazione dei laboratori di genetica. Simona Cavani Maria Isola Parodi Ornella Mazzetti
 L informatizzazione dei laboratori di genetica Simona Cavani Maria Isola Parodi Ornella Mazzetti Laboratorio di Genetica Struttura e patologie analizzate Segreteria / Accettazione Citogenetica -Cariotipo
L informatizzazione dei laboratori di genetica Simona Cavani Maria Isola Parodi Ornella Mazzetti Laboratorio di Genetica Struttura e patologie analizzate Segreteria / Accettazione Citogenetica -Cariotipo
GENETICA E LA SCIENZA CHE STUDIA:
 GENETICA E LA SCIENZA CHE STUDIA: La variabilità biologica degli organismi viventi La trasmissione dei caratteri da un organismo ad un altro o da una cellula ad un altra Il ruolo del genoma (patrimonio
GENETICA E LA SCIENZA CHE STUDIA: La variabilità biologica degli organismi viventi La trasmissione dei caratteri da un organismo ad un altro o da una cellula ad un altra Il ruolo del genoma (patrimonio
28/09/2013 Coop. Medici del Territorio Ara Nova Fiumicino. A.N.M.I. Onlus - Centro Studi Microcitemie Roma
 28/09/2013 Coop. Medici del Territorio Ara Nova Fiumicino screening neonatale (fenilch., fibr. cist., galatt., ipotir. cong.) (esteso: 55 patologie rare) screening prematrimoniale (adulti) diagnosi prenatale
28/09/2013 Coop. Medici del Territorio Ara Nova Fiumicino screening neonatale (fenilch., fibr. cist., galatt., ipotir. cong.) (esteso: 55 patologie rare) screening prematrimoniale (adulti) diagnosi prenatale
Stefano Canali. Talassemie. Storia medica e scientifica. Edizioni ETS
 Stefano Canali Talassemie Storia medica e scientifica Edizioni ETS www.edizioniets.com Volume pubblicato con il contributo di Copyright 2012 EDIZIONI ETS Piazza Carrara, 16-19, I-56126 Pisa [email protected]
Stefano Canali Talassemie Storia medica e scientifica Edizioni ETS www.edizioniets.com Volume pubblicato con il contributo di Copyright 2012 EDIZIONI ETS Piazza Carrara, 16-19, I-56126 Pisa [email protected]
Patologia Molecolare
 Patologia Molecolare MUTAZIONI AD EFFETTO DOMINANTE NEGATIVO DI p53 L effetto fenotipico delle mutazioni da perdita di funzione dipende da: 1) livello residuo di funzione genica 2) soglia di attività necessaria
Patologia Molecolare MUTAZIONI AD EFFETTO DOMINANTE NEGATIVO DI p53 L effetto fenotipico delle mutazioni da perdita di funzione dipende da: 1) livello residuo di funzione genica 2) soglia di attività necessaria
ProCreaMatching. Screening di coppia su geni responsabili per malattie recessive gravi
 ProCreaMatching Screening di coppia su geni responsabili per malattie recessive gravi ProCreaMatching Prevenire le malattie genetiche per difendere la salute del proprio figlio ProCreaLab è un laboratorio
ProCreaMatching Screening di coppia su geni responsabili per malattie recessive gravi ProCreaMatching Prevenire le malattie genetiche per difendere la salute del proprio figlio ProCreaLab è un laboratorio
CORSO INTEGRATO DI GENETICA a.a. 2011-2012. Genetica molecolare in medicina: Analisi di Mutazioni. Cristina Bombieri 7 dicembre 2011
 CORSO INTEGRATO DI GENETICA a.a. 2011-2012 Genetica molecolare in medicina: Analisi di Mutazioni Cristina Bombieri 7 dicembre 2011 GENETICA MOLECOLARE IN MEDICINA SVILUPPI SCIENTIFICI Mappatura gene Identificazione
CORSO INTEGRATO DI GENETICA a.a. 2011-2012 Genetica molecolare in medicina: Analisi di Mutazioni Cristina Bombieri 7 dicembre 2011 GENETICA MOLECOLARE IN MEDICINA SVILUPPI SCIENTIFICI Mappatura gene Identificazione
3 modulo didattico - Le
 3 modulo didattico - Le mutazioni del DNA e le malattie monogeniche. Le mutazioni del genoma umano Mutazione: qualsiasi cambiamento permanente ed ereditabile del DNA Mutazione ereditata proveniente dai
3 modulo didattico - Le mutazioni del DNA e le malattie monogeniche. Le mutazioni del genoma umano Mutazione: qualsiasi cambiamento permanente ed ereditabile del DNA Mutazione ereditata proveniente dai
La mappatura dei geni umani. SCOPO conoscere la localizzazione dei geni per identificarne la struttura e la funzione
 La mappatura dei geni umani SCOPO conoscere la localizzazione dei geni per identificarne la struttura e la funzione Un grande impulso alla costruzione di mappe genetiche è stato dato da le tecniche della
La mappatura dei geni umani SCOPO conoscere la localizzazione dei geni per identificarne la struttura e la funzione Un grande impulso alla costruzione di mappe genetiche è stato dato da le tecniche della
Appunti del corso di Genetica e citogenetica umana
 Anno Accademico 2004-2005 Appunti del corso di Genetica e citogenetica umana Prof.ssa Anna Maria Rossi Corso di studio Scienze Biologiche Molecolari Facoltà di Scienze Matematiche, Fisiche e Naturali Università
Anno Accademico 2004-2005 Appunti del corso di Genetica e citogenetica umana Prof.ssa Anna Maria Rossi Corso di studio Scienze Biologiche Molecolari Facoltà di Scienze Matematiche, Fisiche e Naturali Università
INSIEME, MENO FRAGILI DI PRIMA PROGETTIAMO IL FUTURO
 Il test per la diagnosi molecolare della Sindrome dell'x Fragile: novità e prospettive future Marina Grasso Laboratorio Genetica Umana E.O. Ospedali Galliera 1993-2013 INSIEME, MENO FRAGILI DI PRIMA PROGETTIAMO
Il test per la diagnosi molecolare della Sindrome dell'x Fragile: novità e prospettive future Marina Grasso Laboratorio Genetica Umana E.O. Ospedali Galliera 1993-2013 INSIEME, MENO FRAGILI DI PRIMA PROGETTIAMO
Approccio diagnostico alle sindromi talassemiche
 LE EMOGLOBINOPATIE DIAGNOSI CLINICA E DI LABORATORIO Approccio diagnostico alle sindromi talassemiche F. Manzato (Mantova) 1 The thalassemias constitute a heterogeneous group of naturally occurring, inherit
LE EMOGLOBINOPATIE DIAGNOSI CLINICA E DI LABORATORIO Approccio diagnostico alle sindromi talassemiche F. Manzato (Mantova) 1 The thalassemias constitute a heterogeneous group of naturally occurring, inherit
CORSO INTEGRATO DI GENETICA. a.a /12/2010 Lezioni n Esercizi. Dott.ssa Elisabetta Trabetti
 CORSO INTEGRATO DI GENETICA a.a. 2010-2011 07/12/2010 Lezioni n 51-52 Esercizi Dott.ssa Elisabetta Trabetti (1) Dato il genotipo AaBb: quali sono i geni e quali gli alleli? Disegnate schematicamente questo
CORSO INTEGRATO DI GENETICA a.a. 2010-2011 07/12/2010 Lezioni n 51-52 Esercizi Dott.ssa Elisabetta Trabetti (1) Dato il genotipo AaBb: quali sono i geni e quali gli alleli? Disegnate schematicamente questo
Le mutazioni 2. By NA. Lezione 11
 Le mutazioni 2 Lezione 11 J Sostituzione silente: non determina alcun cambiamento nel prodotto: l amminoacido resta lo stesso. Dal momento che modifica la sequenza puo originare un RFLP. In qualche caso
Le mutazioni 2 Lezione 11 J Sostituzione silente: non determina alcun cambiamento nel prodotto: l amminoacido resta lo stesso. Dal momento che modifica la sequenza puo originare un RFLP. In qualche caso
emoglobinopatie talassemie
 Ematologia (a) Per emoglobinopatie si intendono quelle condizioni morbose, caratterizzate da un alterazione strutturale ereditaria di una delle catene globiniche: le anomalie cliniche associate derivano
Ematologia (a) Per emoglobinopatie si intendono quelle condizioni morbose, caratterizzate da un alterazione strutturale ereditaria di una delle catene globiniche: le anomalie cliniche associate derivano
ASSOCIAZIONE NAZIONALE PER LA LOTTA CONTRO LE MICROCITEMIE IN ITALIA TALASSEMIE ED EMOGLOBINOPATIE
 ASSOCIAZIONE NAZIONALE PER LA LOTTA CONTRO LE MICROCITEMIE IN ITALIA TALASSEMIE ED EMOGLOBINOPATIE Percorsi diagnostici di primo livello. Rassegna di quadri clinici con esercizi e simulazioni condivise
ASSOCIAZIONE NAZIONALE PER LA LOTTA CONTRO LE MICROCITEMIE IN ITALIA TALASSEMIE ED EMOGLOBINOPATIE Percorsi diagnostici di primo livello. Rassegna di quadri clinici con esercizi e simulazioni condivise
La consulenza genetica e la diagnosi prenatale
 X Congresso Nazionale Associazione di Volontariato S.di Cornelia de Lange Gabicce 2-4 novembre 2012 La consulenza genetica e la diagnosi prenatale Dr Angelo Selicorni U.O.S. Genetica Clinica Pediatrica
X Congresso Nazionale Associazione di Volontariato S.di Cornelia de Lange Gabicce 2-4 novembre 2012 La consulenza genetica e la diagnosi prenatale Dr Angelo Selicorni U.O.S. Genetica Clinica Pediatrica
Ringraziamo per l elaborazione dei modelli di refertazione dello schema di Fibrosi Cistica. Ospedali Galliera, Laboratorio di Genetica, Genova
 Roma, 15/02/2011 Viale Regina Elena, 299 00161 Roma Tel: 0649902805 Fax: 0649902292 Email: [email protected] ; [email protected] Gentili colleghi, in fase di valutazione dei risultati delle analisi eseguite
Roma, 15/02/2011 Viale Regina Elena, 299 00161 Roma Tel: 0649902805 Fax: 0649902292 Email: [email protected] ; [email protected] Gentili colleghi, in fase di valutazione dei risultati delle analisi eseguite
Unità Operativa di GENETICA MEDICA. Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE. Responsabile Dott.ssa Alessandra Ferlini
 Unità Operativa di GENETICA MEDICA Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE Responsabile Dott.ssa Alessandra Ferlini Servizi forniti: Definizione genotipica e diagnosi molecolare
Unità Operativa di GENETICA MEDICA Direttore: Prof. Alessandra Ferlini LABORATORIO DI GENETICA MOLECOLARE Responsabile Dott.ssa Alessandra Ferlini Servizi forniti: Definizione genotipica e diagnosi molecolare
SCREENING E DIAGNOSI DELLE ANOMALIE CROMOSOMICHE FETALI
 Ospedale Filippo Del Ponte AZIENDA SOCIO SANITARIA TERRITORIALE DEI SETTE LAGHI S.C. Ginecologia e Ostetricia SCREENING E DIAGNOSI DELLE ANOMALIE CROMOSOMICHE FETALI Informazioni per la paziente Circa
Ospedale Filippo Del Ponte AZIENDA SOCIO SANITARIA TERRITORIALE DEI SETTE LAGHI S.C. Ginecologia e Ostetricia SCREENING E DIAGNOSI DELLE ANOMALIE CROMOSOMICHE FETALI Informazioni per la paziente Circa
FENILCHETONURIA. Adattarsi ad una nuova realtà
 FENILCHETONURIA Adattarsi ad una nuova realtà 9 Che cosa è la fenilchetonuria? Per capire la fenilchetonuria bisogna partire dal concetto che tutti gli alimenti, in quantità variabile, contengono una sostanza
FENILCHETONURIA Adattarsi ad una nuova realtà 9 Che cosa è la fenilchetonuria? Per capire la fenilchetonuria bisogna partire dal concetto che tutti gli alimenti, in quantità variabile, contengono una sostanza
La superficie del globulo rosso è ricca di complesse strutture aventi caratteristiche antigeniche
 La superficie del globulo rosso è ricca di complesse strutture aventi caratteristiche antigeniche galattosio Fucosio N-acetil-glucosamina N-acetil-galattosamina Glicolipide di base eritrocita galattosio
La superficie del globulo rosso è ricca di complesse strutture aventi caratteristiche antigeniche galattosio Fucosio N-acetil-glucosamina N-acetil-galattosamina Glicolipide di base eritrocita galattosio
FEBBRE MEDITERRANEA FAMILIARE: CENNI DI GENETICA
 DAVIDE MARTORANA FEBBRE MEDITERRANEA FAMILIARE: CENNI DI GENETICA PARMA, 23 OTTOBRE 2005 PROBLEMI DELL INQUADRAMENTO DIAGNOSTICO CARATTERISTICA RICORRENTE: EPISODI PERITONITICI ~70% DEI PAZIENTI SUBISCE
DAVIDE MARTORANA FEBBRE MEDITERRANEA FAMILIARE: CENNI DI GENETICA PARMA, 23 OTTOBRE 2005 PROBLEMI DELL INQUADRAMENTO DIAGNOSTICO CARATTERISTICA RICORRENTE: EPISODI PERITONITICI ~70% DEI PAZIENTI SUBISCE
Alberi Genelogici. Le malattie genetiche autosomiche recessive. Mendeliana. Trasmissione ereditaria di un singolo gene. monofattoriale) (eredità
 (eredità") Alberi Genelogici I II SCHEMA DI ALBERI GENEALOGICI 1 2 3 4 1 2 3 4 5 6 7 Trasmissione ereditaria di un singolo gene Mendeliana (eredità monofattoriale) Autosomica Dominante. (AD) III IV 1 2 3 1 4 2 5
Alberi Genelogici I II SCHEMA DI ALBERI GENEALOGICI 1 2 3 4 1 2 3 4 5 6 7 Trasmissione ereditaria di un singolo gene Mendeliana (eredità monofattoriale) Autosomica Dominante. (AD) III IV 1 2 3 1 4 2 5
Diventare genitori è sempre un avventura, e una
 Diventare genitori è sempre un avventura, e una gravidanza è per tutte, in qualche misura, un viaggio verso un mondo di emozioni fortissime e nuove. Quando lungo questa strada si incrocia un compagno di
Diventare genitori è sempre un avventura, e una gravidanza è per tutte, in qualche misura, un viaggio verso un mondo di emozioni fortissime e nuove. Quando lungo questa strada si incrocia un compagno di
LAB-NEWS Anno 1 n 4 Aprile 2006
 1 FAVISMO (DEFICIT G6PD) Cos è il deficit di G6PD? Il deficit di G6PD o favismo è una condizione determinata dalla carenza dell enzima glucosio-6-fosfatodeidrogenasi (G6PD), importante in una via metabolica
1 FAVISMO (DEFICIT G6PD) Cos è il deficit di G6PD? Il deficit di G6PD o favismo è una condizione determinata dalla carenza dell enzima glucosio-6-fosfatodeidrogenasi (G6PD), importante in una via metabolica
PRESTAZIONI SPECIALISTICHE PER LA TUTELA DELLA MATERNITÀ RESPONSABILE, ESCLUSE DALLA PARTECIPAZIONE AL COSTO, IN FUNZIONE PRECONCEZIONALE
 Allegato A PRESTAZIONI SPECIALISTICHE PER LA TUTELA DELLA MATERNITÀ RESPONSABILE, ESCLUSE DALLA PARTECIPAZIONE AL COSTO, IN FUNZIONE PRECONCEZIONALE 1. Prestazioni specialistiche per la donna 89.01 ANAMNESI
Allegato A PRESTAZIONI SPECIALISTICHE PER LA TUTELA DELLA MATERNITÀ RESPONSABILE, ESCLUSE DALLA PARTECIPAZIONE AL COSTO, IN FUNZIONE PRECONCEZIONALE 1. Prestazioni specialistiche per la donna 89.01 ANAMNESI
Polimorfismo clinico e genetico: quali differenze e quali vantaggi
 A.O. Polo Universitario Ospedale L. Sacco, Milano Unità Operativa di Cardiologia Centro Malattie Rare Cardiologiche - Marfan Clinic Responsabile prof. Alessandro Pini Polimorfismo clinico e genetico: quali
A.O. Polo Universitario Ospedale L. Sacco, Milano Unità Operativa di Cardiologia Centro Malattie Rare Cardiologiche - Marfan Clinic Responsabile prof. Alessandro Pini Polimorfismo clinico e genetico: quali
LA GENETICA E L EREDITARIETA
 LA GENETICA E L EREDITARIETA Come si trasmettono i caratteri ereditari? Il primo studioso che osservò le caratteristiche umane e la modalità di trasmissione fu Ippocrate (460-377 a. C.) 1 Come si trasmettono
LA GENETICA E L EREDITARIETA Come si trasmettono i caratteri ereditari? Il primo studioso che osservò le caratteristiche umane e la modalità di trasmissione fu Ippocrate (460-377 a. C.) 1 Come si trasmettono
Formula generale di un amminoacido
 Formula generale di un amminoacido Gruppo carbossilico Gruppo amminico Radicale variabile che caratterizza i singoli amminoacidi Le catene laterali R degli amminoacidi di distinguono in: Apolari o idrofobiche
Formula generale di un amminoacido Gruppo carbossilico Gruppo amminico Radicale variabile che caratterizza i singoli amminoacidi Le catene laterali R degli amminoacidi di distinguono in: Apolari o idrofobiche
Delezione. Duplicazione. Variazioni della struttura. Inversione. Traslocazione. Mutazioni cromosomiche. Nullisomia Monosomia Trisomia Tetrasomia
 Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
Delezione Variazioni della struttura Duplicazione Inversione Mutazioni cromosomiche Variazioni del numero Traslocazione Aneuploidie Nullisomia Monosomia Trisomia Tetrasomia Variazioni del numero di assetti
CARRIER TEST. Scopri se sei portatore sano di malattie genetiche ereditarie
 CARRIER TEST Scopri se sei portatore sano di malattie genetiche ereditarie Che cos è HORIZON HORIZON Carrier Test è un test di screening che permette di valutare se si è portatore sano di uno o più malattie
CARRIER TEST Scopri se sei portatore sano di malattie genetiche ereditarie Che cos è HORIZON HORIZON Carrier Test è un test di screening che permette di valutare se si è portatore sano di uno o più malattie
Classificazione delle anemie
 Emoglobina L emoglobina è una proteina coniugata ad un gruppo prostetico (l eme) essenziale alla funzione di trasporto e cessione di O 2 e CO 2 degli eritrociti. L emoglobina è formata da: 4 Catene globiniche
Emoglobina L emoglobina è una proteina coniugata ad un gruppo prostetico (l eme) essenziale alla funzione di trasporto e cessione di O 2 e CO 2 degli eritrociti. L emoglobina è formata da: 4 Catene globiniche
CORSO INTEGRATO DI GENETICA. a.a /11/2010. Analisi di linkage Analisi di mutazioni
 CORSO INTEGRATO DI GENETICA a.a. 2010-2011 04/11/2010 Lezioni 27_28 Analisi di linkage Analisi di mutazioni Dott.ssa Elisabetta Trabetti LINKAGE Geni in loci vicini sullo stesso cromosoma - concatenati
CORSO INTEGRATO DI GENETICA a.a. 2010-2011 04/11/2010 Lezioni 27_28 Analisi di linkage Analisi di mutazioni Dott.ssa Elisabetta Trabetti LINKAGE Geni in loci vicini sullo stesso cromosoma - concatenati
TEST GENETICI: DEFINIZIONE
 AVVISO Il materiale riportato in queste diapositive è di esclusiva proprietà del Prof. Liborio Stuppia. La pubblicazione delle diapositive su questo sito è intesa unicamente a scopo di consultazione da
AVVISO Il materiale riportato in queste diapositive è di esclusiva proprietà del Prof. Liborio Stuppia. La pubblicazione delle diapositive su questo sito è intesa unicamente a scopo di consultazione da
ANEMIE: ESEMPI PRATICI CASI CLINICI SIMULATI
 ANEMIE: ESEMPI PRATICI CASI CLINICI SIMULATI CASO N. 1 Uomo di 72 anni, lamenta anoressia, calo ponderale di 2 kg in 2 mesi. Hb 8.8 g/dl, GB 3.900/ul, Piastrine 311.000/ul. MCV 75 fl. CASO N. 2 Donna di
ANEMIE: ESEMPI PRATICI CASI CLINICI SIMULATI CASO N. 1 Uomo di 72 anni, lamenta anoressia, calo ponderale di 2 kg in 2 mesi. Hb 8.8 g/dl, GB 3.900/ul, Piastrine 311.000/ul. MCV 75 fl. CASO N. 2 Donna di
Azienda Ospedaliero Universitaria Careggi
 Azienda Ospedaliero Universitaria Careggi Stato dell Arte della misura della HbA 1C Anna Caldini 1 VEQ HbA 1C I Ciclo anno 2002 Materiali in uso Ciclo 2017: Polymed, Emolisati di origine umana, liofilizzati
Azienda Ospedaliero Universitaria Careggi Stato dell Arte della misura della HbA 1C Anna Caldini 1 VEQ HbA 1C I Ciclo anno 2002 Materiali in uso Ciclo 2017: Polymed, Emolisati di origine umana, liofilizzati
Ho pensato di raccogliere in modo organico e chiaro tutte le informazioni pratiche relative alla gravidanza. Mi sono basata sulle emozioni, i dubbi e
 Ho pensato di raccogliere in modo organico e chiaro tutte le informazioni pratiche relative alla gravidanza. Mi sono basata sulle emozioni, i dubbi e gli interrogativi che le future mamme ci hanno espresso
Ho pensato di raccogliere in modo organico e chiaro tutte le informazioni pratiche relative alla gravidanza. Mi sono basata sulle emozioni, i dubbi e gli interrogativi che le future mamme ci hanno espresso
RACCOMANDAZIONI PER LA DIAGNOSTICA DI PRIMO LIVELLO DELLE EMOGLOBINOPATIE della Società Italiana Talassemie ed Emoglobinopatie - SITE
 COLLANA SCIENTIFICA S.I.T.E. N. 1 RACCOMANDAZIONI PER LA DIAGNOSTICA DI PRIMO LIVELLO DELLE EMOGLOBINOPATIE della Società Italiana Talassemie ed Emoglobinopatie - SITE a cura di G. IVALDI e G. BARBERIO
COLLANA SCIENTIFICA S.I.T.E. N. 1 RACCOMANDAZIONI PER LA DIAGNOSTICA DI PRIMO LIVELLO DELLE EMOGLOBINOPATIE della Società Italiana Talassemie ed Emoglobinopatie - SITE a cura di G. IVALDI e G. BARBERIO