Giovanni Monopoli SSFA GdL Dispositivi Medici
|
|
|
- Gabriela Di Pietro
- 8 anni fa
- Visualizzazioni
Transcript
1 1. INTRODUZIONE 2. IL Giovanni Monopoli SSFA GdL Dispositivi Medici 3. CLASSI DI RISCHIO 4. VALUTAZIONE DI CONFORMITÁ E MARCATURA CE 5. BANCA DATI E CLASSIFICAZIONE NAZIONALE I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 2

2 Definizione Esempi Borderline Glossario I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 3 I dispositivi medici sono una categoria in rapida evoluzione che comprende un vasto numero di prodotti (oltre mezzo milione in Europa) molto eterogenei. DEFINIZIONE: si definisce dispositivo medico qualsiasi prodotto (strumento, apparecchio, impianto, sostanza, software o altro) destinato a essere impiegato o a scopo di diagnosi, prevenzione, controllo o terapia, attenuazione o compensazione di ferite o handicap, ma anche di studio, sostituzione o modifica o di un processo fisiologico, o di controllo del concepimento. Esso svolge la sua funzione attraverso una modalitá che non é farmacologica, immunologica o metabolica, pur potendo essere coadiuvato da una o piú di tali modalitá (azione ancillare). La destinazione del prodotto deve essere in ogni caso connotabile come finalitá medica. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 4

3 Termometri, cerotti, apparecchi radiografici, valvole cardiache, stent coronarici, protesi articolari, profilattici, abbassalingua, lacci emostatici, guanti, siringhe, occhiali graduati... identificazione di un prodotto nella specifica categoria di appartenenza é molto importante al fine di individuare quale sia la normativa di riferimento da applicare. Essa non é sempre facile. Esistono prodotti borderline, che per loro natura non appartengono con chiarezza ad un determinato settore, per cui su tali prodotti si possono applicare altre normative di riferimento. In alcuni casi dubbi che si possono prestare ad interpretazioni diverse della definizione, la destinazione del DM, che deve in ogni caso essere connotabile con una finalitá medica, é utile ad escludere da tale categoria alcuni prodotti (cosmetici, erboristici, integratori alimentari, apparecchiature estetiche). La differenza fondamentale con le sostanze medicinali (tenendo conto che lo scopo é sovrapponibile e che un DM puó anche essere una sostanza) risiede nel fatto che il medicinale agisce per definizione (D. Lgs. 219/2006) con mezzi farmacologici, metabolici o immunologici. principale di un DM si esplica con mezzi fisici (azione meccanica, lavaggio, sostituzione o supporto per organi o funzioni corporee, etc.). Spetta sempre al fabbricante decidere se il proprio prodotto rientra nella definizione di DM. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 5 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 6

4 Fabbricante: la persona fisica o giuridica responsabile della progettazone, fabbricazione, imballaggio ed etichettatura di un DM al fine in commercio a proprio nome, indipendentemente dal fatto che queste operazioni siano eseguite da questa stessa persona o da un terzo per suo conto Mandatario: la persona fisica o giuridica stabilita nel territorio Europea che, dopo essere stata espressamente designata dal fabbricante, agisce e puó essere interpellata dalle autoritá nazionali competenti e dagli organismi comunitari in vece del fabbricante per quanto riguarda gli obblighi di legge Destinazione: alla quale il DM é destinato secondo le indicazioni fornite dal fabbricante nel foglio illustrativo o nel materiale pubblicitario Dispositivo Medico Attivo: qualsiasi DM collegato per il suo funzionamento ad una fonte di energia elettrica o a qualsiasi altra fonte di energia diversa da quella prodotta dal corpo umano o dalla gravitá Dispositivo Medico Impiantabile Attivo: qualsiasi DM attivo destinato ad essere impiantato interamente o parzialmente mediante intervento chirurgico o medico nel corpo umano o mediante intervento medico in un orifizio naturale e destinato a restarvi dopo I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 7 Dispositivo Medico-Diagnostico in Vitro (IVD): qualsiasi DM composto da un reagente, un reattivo, un calibratore, un materiale di controllo, un kit, uno strumento, un apparecchio, o un sistema, usato da solo o in combinazione, destinato dal fabbricante ad essere impiegato in vitro per di campioni provenienti dal corpo umano, iclusi sangue e tessuti donati, unicamente o principalmente allo scopo di fornire informazioni su uno stato fisiologico o patologico, o su una anomalia congenita, o infomazioni che consentono la determinazione della sicurezza e della compatibilitá con potenziali soggetti riceventi, o che consentono il controllo delle misure terapeutiche Categoria con caratteristiche differenti dagli altri DM e piú omogenea, che viene trattata separatamente I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 8

5 Riferimenti Principi del I requisiti essenziali Il dossier tecnico Norme tecniche armonizzate dei rischi ed i sistemi di qualitá La Direttiva 2007/47/CE I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 9 Normativa Titolo/Argomento Pubblicazione DLgs n. 507 del 14/12/1992 DLgs n. 46 del 24/02/1997 DLgs n /09/2000 DLgs n. 37 del 25/01/2010 D.M. 02/08/2005 Nota di chiarimento 26/02/2007 Nota di chiarimento 05/12/2007 D.M. 15/11/2005 Attuazione direttiva 90/385/CEE relativa ai dispositivi medici impiantabili attivi G.U. 30/12/1992, n. 305 Attuazione direttiva 93/42/CEE concernente i dispositivi G.U. 6/03/1997, n. 54 medici Attuazione direttiva 98/79/CE relativa ai dispositivi G.U. 17/09/2000, n. 269 medico-diagnostici in vitro Attuazione della direttiva 2007/47/CE che modifica le direttive 90/385/CEE, 93/42/CEE e 98/8/CE Modalità di presentazione della documentazione per la notifica di indagine clinica con dispositivi medici Indagini cliniche post-marketing (dispositivi recanti marchio CE e posti in commercio) Modalità di presentazione della documentazione da inoltrare per la notifica di indagini cliniche e chiarimenti alla Nota 26/2/2007 G.U. 13/03/2010, n. 60 G.U. 09/09/2005, n. 210 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 10 N.A. N.A. Approvazione modelli di schede segnalazione incidenti G.U. 24/11/2005, n. 274 relativi a dispositivi medici o dispositivi medicodiagnostici in vitro Linee Guida MEDDEV: documenti non vincolanti elaborati a seguito di consultazione a livello comunitario tra le varie parte interessate (AC, Commissione Europea, industria, etc) e ne rappresentano la posizione condivisa

6 in commercio dei DM é regolamentata su base comunitaria e il sistema adottato viene definito (adottato per la prima volta nel 1985 al fine di rimuovere ostacoli tecnici agli scambi nel mercato interno), un sistema di certificazione e garanzia differente dal sistema autorizzativo/registrativo in uso p.es. per i farmaci. Sistema autorizzativo Nuovo approccio I principi fondamentali del nuovo approccio sono: assenza di una autorizzazione preventiva per in commercio da parte delle Autorità Competenti (e obbligo della comunicazione Competente per la commercializzazione) conformitá dei prodotti ai requisiti essenziali previsti dalle direttive valutazione della conformità dei prodotti da parte del fabbricante (e, nei casi previsti, da parte di un organismo notificato) sorveglianza del mercato da parte delle Autorità Competenti libera circolazione dei prodotti nei Paesi La conformitá ai requisiti essenziali é dimostrata dalla presenza sul prodotto del marchio CE e da parte del fabbricante, della dichiarazione di conformitá alla/e direttiva/e applicabile/i I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 11 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 12
 conformitá dei prodotti ai requisiti essenziali previsti dalle direttive valutazione della conformità dei prodotti da parte del fabbricante (e,")
7 I requisiti essenziali dei DM e dei DMIA (DM Impiantabili Attivi) sono descritti negli allegati delle rispettive direttive e sono suddivisi in: Requisiti generali: riguardano sicurezza e prestazioni intrinseche del dispositivo e applicabili a tutti i prodotti (sicurezza e salute di pazienti ed utilizzatori, analisi dei rischi e minimizzazione dei rischi garanzia delle prestazioni, inalterabilitá delle caratteristiche, valutazione clinica) Requisiti relativi alla progettazione e costruzione: riguardano gli aspetti tecnologici e sono applicabili a tipologie di prodotto (caratteristiche chimiche, fisiche, meccaniche e biologiche, infezione e contaminazione microbica, caratteristiche relative alla costruzione ed caratteristiche relative a dispositivi con funzione di misura, radiazioni, fonte di energia, informazioni fornite dal fabbricante) Ciascun DM deve essere corredato dalle informazioni che garantiscano la sua identificazione, destinazione e modalitá per una utilizzazione sicura. Tale garanzia costituisce un requisito essenziale. Tali indicazioni sono riportate in etichetta e sulle istruzioni per in lingua italiana. Esse devono figurare quando possibile sul DM e. Se unitario non é fattibile, le istruzioni devono figurare su un foglio illustrativo. deve contenere: nome/ragione sociale e indirizzo fabbricante identificazione del DM e contenuto, tipologia del DM condizioni di conservazione e/o manipolazione istruzioni specifiche, avvertenze e/o precauzioni del caso e/o monouso e/o dispositivo su misura e/o destinato esclusivamente ad indagini cliniche lotto e data entro cui il DM dovrebbe essere utilizzato I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 13 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 14

8 Le istruzioni per possono essere riportate sul foglietto illustrativo o sul manuale e possono essere omesse solo per i DM meno critici. Esse devono riportare: indicazioni (tranne data di scadenza /fabbricazione e lotto) prestazioni ed effetti collaterali informazioni sulla connsessione con altri DM informazioni su installazione e manutenzione rischi connessi con o di interferenze trattamenti appropriati informazioni per i DM che emettono radiazioni indicazioni che permettano agli operatori sanitari di informare adeguatamente i pazienti in merito a controindicazioni o precauzioni (esposizione a campi magnetici, eliminazione, medicinali contenuti nel DM o che il DM puó somministrare, etc). con la direttiva 2007/47/CE é stata aggiunta la data di emissione versione delle istruzioni I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 15 É compito del fabbricante accertare la rispondenza del prodotto ai requisiti tecnici ad esso aplicabili, documentando tale processo in un dossier tecnico. Le direttive elencano la documentazione che deve essere inclusa nel dossier tecnico (relativa a: progettazione, gestione dei rischi, fabbricazione, etichette ed istruzioni per dati clinici, sorveglianza post-marketing, norme tecniche applicate, etc). I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 16
.")
9 Il dossier tecnico deve possedere la seguente struttura minima: Descrizione del prodotto Schema di progettazione e metodi di fabbricazione Risultati del rischio Norme tecniche applicate Tabella di rispondenza ai requisiti essenziali Relazioni di prova e dati clinici Progetto di etichettatura ed eventualmente di istruzioni Dato il rapido avanzamento tecnologico del settore, le procedure alle quali il fabbricante deve fare riferimento per raggiungere ai requisiti tecnici non sono inserite nelle direttive (ció evita la rapida obsolescenza delle direttive stesse). Le specifiche tecniche sono invece contenute nelle norme tecniche armonizzate, elaborate da organismi riconosciuti a svolgere attivitá normativa. Norma Internazionale (ISO International Organization for Standardization) Norma Europea (CEN Comitée Europeen de Normalisation) Norma Nazionale (UNI Ente Nazionale Italiano di Unificazione) Esse sono consensuali, volontarie e democratiche e riflettono lo stato relativamente alle conoscenze nel settore. Le norme tecniche si dicono armonizzate quando sono adottate a livello europeo, su mandato della Commissione Europea, dai Comitati Europei di Normalizzazione (CEN) e pubblicate come elenco nella Gazzetta ufficiale Europea. Solitamente a livello europeo si adottano Norme Internazionali con eventuali adattamenti. Il loro rispetto assicura, come espressamente riportato in tutte le direttive del nuovo approccio, una presunzione di conformitá (purché una norma tecnica sia applicata integralmente). Nel caso non fossero utilizzate, il fabbricante é tenuto a dimostrare interamente tale conformitá. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 17 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 18

10 Recepita in Italia tramite Decreto Legislativo del 25 Gennaio 2010, n. 37. Le principali novità riguardano i seguenti aspetti: Obbligo per i fabbricanti alla valutazione clinica (letteratura e/o indagini cliniche): precedentemente prevista solo per DM a maggior rischio Comunicazioni su rifiuto o interruzione di sperimentazioni cliniche Aggiornamento post-marketing della valutazione clinica Intervento EMA per dispositivi che incorporano una sostanza medicinale Introduzione e definizione del DM stand (attivo) Precisazione del concetto di Riclassificazione di alcuni DM in una classe di rischio piú alta Validazione del software dei DM attivi Disposizioni per avvio definitivo della banca dati europea EUDAMED Obbligo di conservazione della documentazione prolungato per alcune tipologie di dispositivi (impiantabili) fino a 15 anni Armonizzazione normativa vigilanza e sistema sanzionatorio Classificazione dei dispositivi medici Classi di rischio Regole di classificazione I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 19 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 20
 Precisazione del concetto di Riclassificazione di alcuni DM in una classe di rischio piú alta Validazione del software dei DM attivi Disposizioni per")
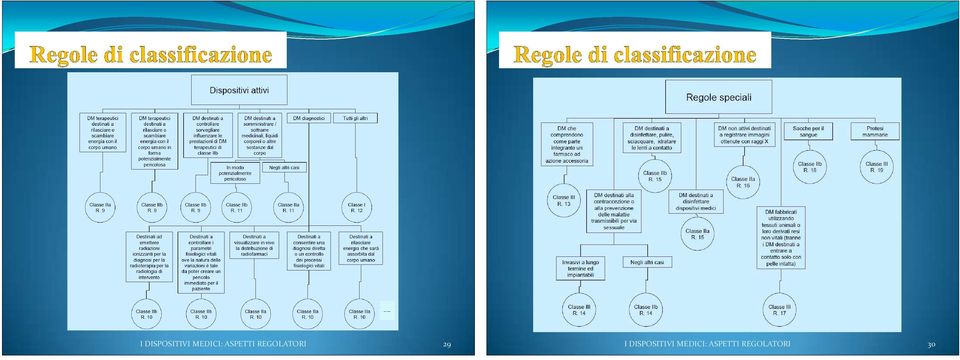
11 Il fabbricante deve classificare il proprio DM per poter eseguire la valutazione di conformitá ai requisiti essenziali (le procedure differiscono a seconda della classe di appartenenza). I DM sono classificati in classi di rischio I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 21 Classe I: dispositivi meno critici quali la gran parte di quelli non attivi e non invasivi (poltrone dentistiche, lenzuola monouso) Classe Is: dispositivi di classe I forniti allo stato sterile (cerotti, bende) Classe Im: dispositivi di classe I che svolgono una funzione di misura (misuratori di pressione sanguigna non automatizzati, bilance mediche) Classe IIa: dispositivi a rischio medio. Alcuni dispositivi non attivi (invasivi e non) e i dispositivi attivi che interagiscono con il corpo in maniera non pericolosa (apparecchi acustici, lenti a contatto) Classe IIb: dispositivi a rischio medio/alto. Alcuni dispositivi non attivi (invasivi e non) e la maggior parte dei dispositivi che interagiscono con il corpo (defibrillatori, incubatori per neonati) Classe III: dispositivi ad alto rischio, quali gran parte dei dispositivi impiantabili, quelli contenenti farmaci o derivati animali, ed alcuni dispositivi che interagiscono sulle funzioni di organi vitali (dispositivi per infusione e trasfusione di sangue, apparecchi per emodialisi, cardiostimolatori) I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 22
 Classe IIa: dispositivi a rischio medio.")
 dalla sede anatomica su cui incide il dispositivo (in")
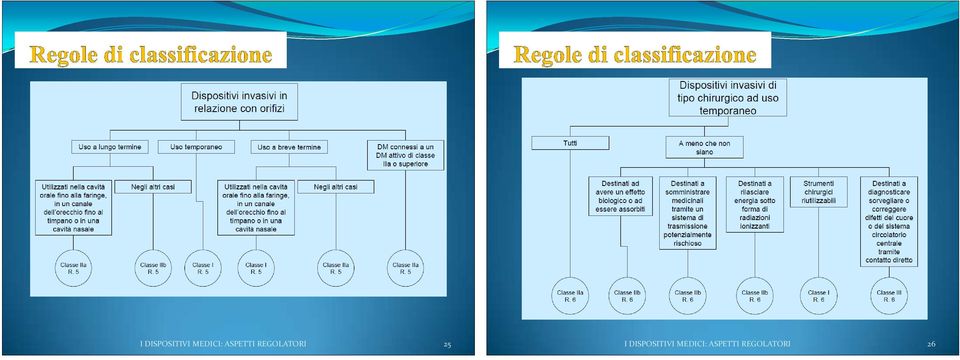
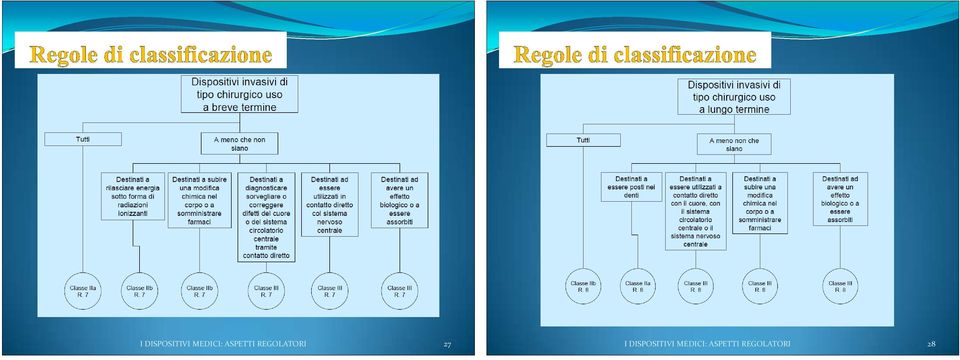
12 I criteri e le regole di classificazione dipendono: dalla invasivitá (dispositivi non invasivi, invasivi negli orifizi del corpo, invasivi chirurgici, impiantabili); dalla durata* del contatto del dispositivo con il paziente dal tipo di funzionamento (dispositivo non attivo, dispositivo attivo terapeutico, dispositivo attivo diagnostico) dalla sede anatomica su cui incide il dispositivo (in particolare sistema circolatorio centrale e sistema nervoso centrale ) *Durata: Temporanea : inferiore a 60 minuti Breve termine : inferiore a 30 giorni Lungo termine : superiore a 30 giorni I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 23 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 24


13 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 25 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 26

14 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 27 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 28

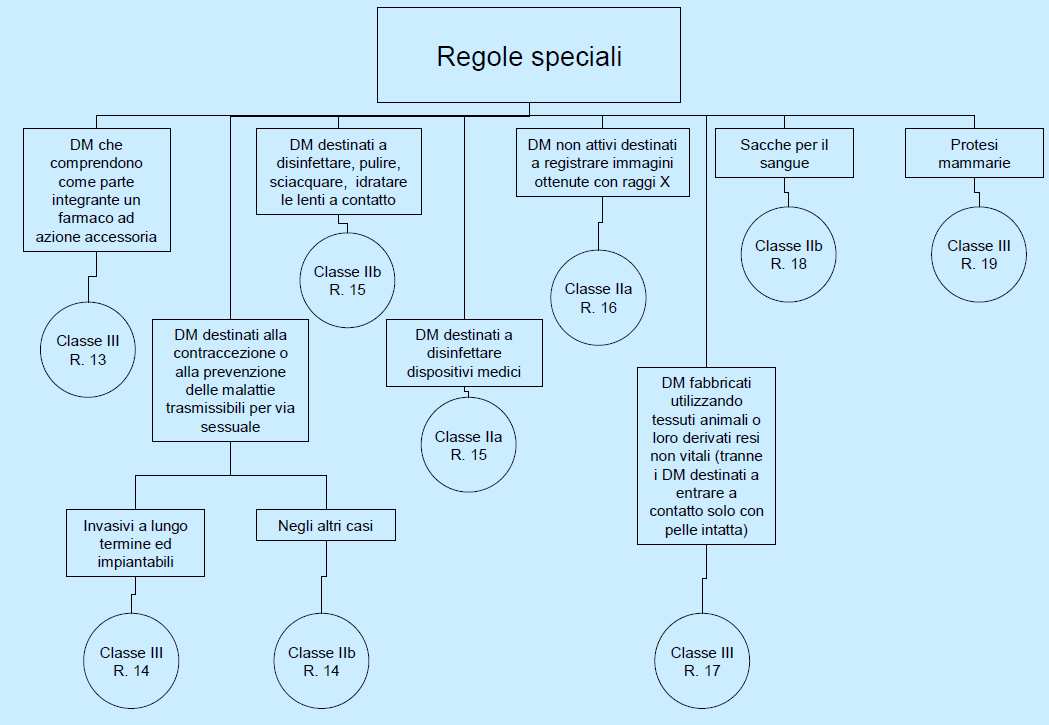
15 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 29 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 30
16 I DMIA rappresentano una tipologia di prodotti omogenea, soprattutto riguardo al funzionamento, per cui la direttiva che li regolamenta non prevede una distinzione in classi di rischio. A tutti i DMIA (compresi i loro accessori, anche se non impiantabili o non attivi) si applicano le stesse disposizioni previste dalla direttiva stessa. La MEDDEV 2.4/1-rev 8 Part 1-2 (Luglio 2001) offre una guida di tali regole, riportando esempi specifici. Tra le modifiche introdotte a seguito della direttiva 2007/47/CE e della direttiva 2005/50/CE: DM a contatto diretto con riclassificati in classe III, a seguito della modifica della definizione di sistema circolatorio centrale introdotta dalla direttiva Le protesi spalla e ginocchio sono state riclassificate in classe III, in deroga alle regole di classificazione (che le collocavano in classe IIb), al fine di consentire una piú approfondita valutazione della loro progettazione da parte ON Attestazione della conformitá Organismi Notificati Procedure per la valutazione della conformitá Scelta della procedura Intervento Notificato Procedura per i Dispositivi Medici Impiantabilli Attivi Marcatura CE I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 31 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 32

17 I documenti che attestano la conformitá di un DM alle disposizioni della direttiva applicabile e che ne consentono la marcatura CE e in commercio sono la Dichiarazione di Conformitá ed il Certificato CE. La conformitá viene valutata per tutti i DM dal fabbricante, che la attesta mediante la Dichiarazione di Conformitá. Per i DM di classe superiore alla I e per alcuni aspetti relativi ai DM di classe Is e Im, la conformitá viene valutata, con modalitá diverse, anche da un soggetto terzo, detto Organismo Notificato, che la attesta rilasciando il Certificato CE al fabbricante. DICHIARAZIONE DI CONFORMITÁ Documento con il quale il fabbricante garantisce e dichiara che i propri prodotti soddisfano le disposizioni applicabili della direttiva di riferimento. Essa costituisce di responsabilitá da parte del fabbricante. Le direttive riportano le informazioni necessarie nella dichiarazione solo in maniera generica. La norma tecnica EN fornisce criteri generali per la sua preparazione. CERTIFICATO CE Documento con il quale una terza parte (Organismo Notificato) certifica di aver svolto un processo di Valutazione della rispondenza di un prodotto alle disposizioni applicabili della direttiva di riferimento ORGANISMO NOTIFICATO (ON) Un fabbricante puó richiedere la certificazione ad un ON autorizzato da una qualsiasi Autoritá Competente. La certificazione consente la marcatura CE del prodotto e la sua libera circolazione nei paesi comunitari. (documentale ed ispettiva); rinnovabile alla scadenza previa rivalutazione del Al momento sono autorizzati in Italia dieci OONN ai sensi del D.Lgs. 46/97 (DM), mentre per la certificazione dei DMIA un solo ON é autorizzato (ISS). I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 33 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 34

18 completo degli OONN italiani é presente nel sito internet del Ministero della Salute, area tematica dispositivi medici: No I.M.Q. S.p.A. No ISTITUTO DI RICERCA E COLLAUDI M. MASINI No ISTITUTO SUPERIORE DI SANITA' No C.P.M. ISTITUTO RICERCHE PROVE ANALISI S.r.l. No ICIM Istituto di Certificazione Industriale per la Meccanica S.p.A. No ITALCERT No CERMET Soc. Cons. r.l. No BIOLAB No CERTIQUALITY No TÜV Rheinland Italia srl Requisiti che un Ente (e le strutture eventualmente utilizzate da tale Ente) deve possedere per essere autorizzato come ON: personale ed apparecchiature adeguate, manuale di qualitá, copertura economica di bilancio ed assicurativa, locali idonei e a norma, requisiti di indipendenza, imparzialitá, riservatezza, integritá professionale e competenza. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 35 La valutazione di conformitá di un prodotto ai requisiti essenziali previsti da ciascuna direttiva di nuovo approccio si esegue utilizzando moduli ad hoc stabiliti per Decisione del Consiglio dei Ministri CEE. La procedura varia secondo il rischio e la conseguente classificazione del prodotto. La valutazione di conformitá deve riguardare tutti gli aspetti del prodotto, relativi sia alla progettazione che alla fabbricazione. I moduli di base sono otto e possono essere combinati tra loro in diversi modi al fine di definire procedure complete di valutazione di conformitá. I moduli C e G non sono applicati ai DM I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 36
 deve possedere per essere autorizzato come ON: personale ed apparecchiature adeguate,")
19 La direttiva dei DM (93/42/CE) ha adattato sei di questi moduli negli allegati II VII secondo il seguente schema: ALLEGATO II = MODULO H ALLEGATO III = MODULO B ALLEGATO IV = MODULO F ALLEGATO V = MODULO D ALLEGATO VI = MODULO E ALLEGATO VII = MODULO A Ciascun allegato riporta la documentazione che il fabbricante deve approntare, gli adempimenti che deve attuare le eventuali dichiarazioni di conformitá che deve redigere e certificazioni che deve richiedere. La direttiva riporta inoltre per ciascuna classe di DM gli allegati applicabili, le loro eventuali possibili combinazioni e le opzioni di scelta del fabbricante. La scelta delle procedure per certificare un DM di una medesima classe dipende esclusivamente dal fabbricante che opererà tale scelta sulla base della propria organizzazione (per esempio presenza o meno di un sistema di qualità) o in base al tipo, numero o frequenza della produzione dei propri DM. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 37 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 38

20 La diversitá delle procedure per la valutazione di conformitá e la diversa applicabilitá degli allegati dipendono in particolare o meno e dalla sua tipologia: CLASSE I: intervento non é previsto CLASSE Is e Im: la valutazione riguarda solo alcuni aspetti della produzione (quelli legati alla sterilizzazione per i dispositivi di classe Is e quelli legati agli aspetti metrologici per i dispositivi di classe Im) CLASSE IIa: la valutazione riguarda solo gli aspetti della produzione CLASSE III E IIb: la valutazione riguarda tutti gli aspetti connessi al DM che vanno dalla progettazione alla produzione ON puó essere di due tipi specifici: Certificazione di prodotto Verifica della progettazione di un esemplare tipo (All. III) Verifica di un campione di un lotto di produzione (All. IV) Certificazione dei sistemi di qualitá Sistema di qualitá completo: progettazione, produzione, prove finali (All. II) Sistema di qualitá della produzione (All. V) Sistema di qualitá della produzione limitato alle prove finali (All. VI) La Direttiva 2007/47/CE ha introdotto la necessità di verifica da parte ON di un certo numero di dossier tecnici (proporzionale alla classe, IIa o IIb) anche quando sono applicati i moduli relativi ai sistemi di qualità I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 39 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 40

.")
21 Le procedure di valutazione dei DMIA sono le stesse per tutte le tipologie di prodotto (non sono distinti in classi diverse). Tali procedure sono equiparabili a quelle previste per i DM di classe III (piú alto rischio). Gli allegati riportati nella direttiva dei DMIA corrispondono a quelli dei DM applicabili alla classe III ma sono indicati con numeri arabi anziché romani: Allegato 2 (compreseo della progettazione) Allegati oppure Allegati Unico ON autorizzato in Italia per i DMIA: ISS Quando il fabbricante é in possesso della dichiarazione di conformita ed eventualmente del certificato rilasciato (o dei certificati), puó marcare CE il proprio DM e immetterlo in commercio. Il marchio CE deve essere apposto in maniera visibile, leggibile ed indelebile sul DM o sterile e, se del caso, anche sulla confezione commerciale e sulle istruzioni per. In caso di intervento il marchio deve essere corredato dal numero di codice identificativo stesso. La documentazione tecnica e i documenti di attestazione della conformitá devono essere conservati dal fabbricante o mandatario per almeno cinque anni dalla data di frabbricazione DM. Per i dispositivi impiantabili, tale periodo é stato esteso ad almeno quindici anni dalla data di fabbricazione DM con la direttiva 2007/47/CE. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 41 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 42
22 La Banca Dati Nazionale (BD) Il Repertorio dei Dispositivi Medici Classificazione Nazionale dei Dispositivi Medici (CND) Al fine di assicurare efficaci collegamenti tra le Autoritá Competenti degli Stati membri, é stata prevista di una Banca Dati Europea (EUDAMED). Essa non é ancora pienamente operante e (da attivare entro il 5 Settembre 2012) verrá alimentata dagli Stati Membri attraverso delle informazioni sui DM raccolte dagli stessi e conterrá anche dati relativi alle indagini cliniche. Ogni Stato deve dunque dotarsi di un sistema nazionale di comunicazione dei DM. A tal proposito il Ministero della Salute ha realizzato la Banca Dati nazionale (BD), dove fabbricanti di DM (con sede in Italia e nei Paesi dell UE) devono registrarsi obbligatoriamente. Con il D. Lgs 25 Gennaio 2010 n. 37 di recepimento della Direttiva anche per i DMIA nella BD (inoltre, per i DM di classe I é stato previsto stabilita sul territorio italiano) I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 43 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 44
23 La registrazione dei dati avviene in formato elettronico, attraverso il web ed in nessun caso configura alcuna forma di autorizzazione da parte del Ministero della Salute. I dati inseriti devono essere validati mediante apposizione della Firma Digitale. Dati richiesti: caratteristiche tecniche generali, materiali, eventuali modalitá di sterilizzazione, presenza di tessuti o sostanze di origine animale o di medicinali o derivati del sangue, invio in formato elettronico e delle istruzioni per. Per i soggetti che assemblano DM valgono le disposizioni previste per fabbricanti e mandatari di DM di classe I. Per i dispositivi su misura tali disposizioni non si applicano e resta in vigore di comunicazione dei dati per via cartacea. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 45 In questo quadro, in ambito nazionale, si inserisce la legge finanziaria del 2003 (e aggiornamenti della legge finanziaria del 2006), che istituisce presso il Ministero della Salute, la Commissione Unica dei Dispositivi medici (CUD), con il compito di definire e aggiornare il Repertorio dei DM. Pertanto, della BD é stata inserita la facoltá di registrare i DM nel Repertorio, dichiarando la disponibilitá alla vendita degli stessi al Servizio Sanitario Nazionale. Il repertorio si configura dunque come sottoinsieme di informazioni della BD e la registrazione in entrambi i sistemi si esegue con operazione (numero di iscrizione al repertorio = numero iscrizione alla BD con suffisso /R monitoraggio dei consumi dei DM acquistati dal SSN (finanziaria 2006) e verifica da parte del SSN ottemperanza obblighi di comunicazione fabbricanti. I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 46
24 Normativa Titolo/Argomento Pubblicazione D.M. 20/02/2007 Approvazione della Classificazione G.U. 16/03/2007, n. 63 Nazionale dei Dispositivi Medici D.M. 13/03/2008 Modifiche alla Classificazione Nazionale G.U. 29/05/2008, n. 125 dei Dispositivi Medici D.M. 12/02/2010 Modifiche ed aggiornamenti alla G.U. 24/05/2010, n. 119 Classificazione nazionale dei dispositivi medici (CND), di cui al decreto del Ministero della salute 20 febbraio 2007 La legge finanziaria del 2003 prevedeva che il CUD procedesse anche a classificare tutti i prodotti in classi e sottoclassi specifiche con delprezzodiriferimento La CND permette di raggruppare i dispositivi in categorie omogenee di prodotti (dispositivi destinati ad effettuare un intervento diagnostico o terapeutico simile) e contiene anche i DMIA. La CND ha una struttura ad albero ramificato ed é costituita da: categorie, gruppi e tipologie. Nella seconda versione della CND é stata inserita la categoria W degli IVD (Totale: 22 categorie, 21 gruppi e 3120 rami dei quali 1802 relativi a IVD) Categorie dei Descrizione Dispositivi Medici A DISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA B DISPOSITIVI PER EMOTRASFUSIONE ED EMATOLOGIA C DISPOSITIVI PER APPARATO CARDIOCIRCOLATORIO D DISINFETTANTI, ANTISETTICI E PROTEOLITICI (D. Lgs. 46/97) F DISPOSITIVI PER DIALISI G DISPOSITIVI PER APPARATO GASTROINTESTINALE H DISPOSITIVI DA SUTURA J DISPOSITIVI IMPIANTABILI ATTIVI K DISPOSITIVI PER CHIRURGIA MINI-INVASIVA ED ELETTROCHIRURGIA L STRUMENTARIO CHIRURGICO PLURIUSO O RIUSABILE M DISPOSITIVI PER MEDICAZIONI GENERALI E SPECIALISTICHE N DISPOSITIVI PER SISTEMA NERVOSO E MIDOLLARE P DISPOSITIVI PROTESICI IMPIANTABILI E PRODOTTI PER OSTEOSINTESI Q DISPOSITIVI PER ODONTOIATRIA, OFTALMOLOGIA E OTORINOLARINGOIATRIA R DISPOSITIVI PER APPARATO RESPIRATORIO E ANESTESIA S PRODOTTI PER STERILIZZAZIONE T DISPOSITIVI DI PROTEZIONE E AUSILI PER INCONTINENZA (D. Lgs. 46/97) U DISPOSITIVI PER APPARATO UROGENITALE V DISPOSITIVI VARI W DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO (D. Lgs. 332/2000) Y SUPPORTI O AUSILI TECNICI PER PERSONE DISABILI Z APPARECCHIATURE SANITARIE E RELATIVI COMPONENTI ACCESSORI E MATERIALI I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 47 I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 48
25 Codice ramo CND A A01 A0101 A0102 A0103 A0104 A0105 A0106 A0180 A0190 A0199 A02 A0201 A0202 A0280 A0299 Descrizione ramo CND DISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA AGHI AGHI E KIT PER INFUSIONE E PRELIEVO AGHI E KIT PER BIOPSIA AGHI E KIT PER ANESTESIA AGHI PER DIALISI AGHI E KIT PER INIEZIONE IN OFTALMOLOGIA AGHI E KIT PER ODONTOIATRIA AGHI - ACCESSORI AGHI PER PROCEDURE VARIE AGHI - ALTRI SIRINGHE SIRINGHE MONOUSO SIRINGHE PLURIUSO SIRINGHE - ACCESSORI SIRINGHE - ALTRE I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 49 gmonopoli@clintec.com I DISPOSITIVI MEDICI: ASPETTI REGOLATORI 50
PROCEDURA OPERATIVA PER LA GESTIONE
 Proc. 19 Pag. 1 di 7 PROCEDURA OPERATIVA PER LA GESTIONE DEGLI ADEMPIMENTI DELLA DIRETTIVA 93/42/CEE CONCERNENTE I DISPOSITIVI MEDICI 1. SCOPO... 2 2. APPLICABILITÀ... 2 3. DOCUMENTI DI RIFERIMENTO...
Proc. 19 Pag. 1 di 7 PROCEDURA OPERATIVA PER LA GESTIONE DEGLI ADEMPIMENTI DELLA DIRETTIVA 93/42/CEE CONCERNENTE I DISPOSITIVI MEDICI 1. SCOPO... 2 2. APPLICABILITÀ... 2 3. DOCUMENTI DI RIFERIMENTO...
Il Punto di vista dell Organismo notificato
 Il Punto di vista dell Organismo notificato II Conferenza Nazionale dei Dispositivi Medici 17/19 Marzo 2008 - Villa Erba - Cernobbio Relatore: Gabriele Lualdi - Istituto di Ricerche e Collaudi M. Masini
Il Punto di vista dell Organismo notificato II Conferenza Nazionale dei Dispositivi Medici 17/19 Marzo 2008 - Villa Erba - Cernobbio Relatore: Gabriele Lualdi - Istituto di Ricerche e Collaudi M. Masini
MARCATURA CE E ISO 13485 NEL SETTORE DEI MEDICAL DEVICES. Sara Pelizzoli. Modena, 29/10/2014. - Copyright Bureau Veritas
 MARCATURA CE E ISO 13485 NEL SETTORE DEI MEDICAL DEVICES Sara Pelizzoli Modena, 29/10/2014 - Copyright Bureau Veritas RIFERIMENTI NORMATIVI Direttiva 93/42/CEE del Consiglio del 14 Giugno 1993 concernente
MARCATURA CE E ISO 13485 NEL SETTORE DEI MEDICAL DEVICES Sara Pelizzoli Modena, 29/10/2014 - Copyright Bureau Veritas RIFERIMENTI NORMATIVI Direttiva 93/42/CEE del Consiglio del 14 Giugno 1993 concernente
DATASOFTWARE s.r.l. Soluzioni Informatiche per le Ortopedie e Sanitarie
 I DISPOSITIVI MEDICI Sono una categoria di prodotti (strumenti, apparecchi, impianti, sostanze, software o altro) destinati ad essere impiegati nell uomo o sull uomo a scopo di diagnosi, prevenzione, controllo
I DISPOSITIVI MEDICI Sono una categoria di prodotti (strumenti, apparecchi, impianti, sostanze, software o altro) destinati ad essere impiegati nell uomo o sull uomo a scopo di diagnosi, prevenzione, controllo
Sistema europeo di regolamentazione per i dispositivi medici
 Sistema europeo di regolamentazione per i dispositivi medici Il campo di gioco Nuovo Approccio all'armonizzazione tecnica e Approccio Globale alla valutazione della conformità L'articolo 95 paragrafo 3
Sistema europeo di regolamentazione per i dispositivi medici Il campo di gioco Nuovo Approccio all'armonizzazione tecnica e Approccio Globale alla valutazione della conformità L'articolo 95 paragrafo 3
DISPOSITIVI. Federico II. Avv. Antonia De Lisio
 DISPOSITIVI Le direttive comunitarie e le norme italiane che hanno recepito le direttive stesse disciplinano, separatamente, tre categorie di dispositivi medici 1 DISPOSITIVI i dispositivi medici impiantabili
DISPOSITIVI Le direttive comunitarie e le norme italiane che hanno recepito le direttive stesse disciplinano, separatamente, tre categorie di dispositivi medici 1 DISPOSITIVI i dispositivi medici impiantabili
Incontro con ASSOBIOMEDICA Roma, 07 maggio 2012
 Il sistema BD/RDM: integrazione dei Dispositivi medico diagnostici in vitro La rilevazione degli IVD Incontro con ASSOBIOMEDICA Roma, 07 maggio 2012 Agenda Le attuali modalità di notifica per i dispositivi
Il sistema BD/RDM: integrazione dei Dispositivi medico diagnostici in vitro La rilevazione degli IVD Incontro con ASSOBIOMEDICA Roma, 07 maggio 2012 Agenda Le attuali modalità di notifica per i dispositivi
MARCATURA CE DELLE APPARECCHIATURE PER RADIOLOGIA MEDICA CONFORMI ALLA DIRETTIVA 93/42/CEE
 MARCATURA CE DELLE APPARECCHIATURE PER RADIOLOGIA MEDICA CONFORMI ALLA DIRETTIVA 93/42/CEE DIRETTIVE COMUNITARIE Disposizioni rivolte ai governi degli Stati membri, i quali hanno l obbligo di recepirle
MARCATURA CE DELLE APPARECCHIATURE PER RADIOLOGIA MEDICA CONFORMI ALLA DIRETTIVA 93/42/CEE DIRETTIVE COMUNITARIE Disposizioni rivolte ai governi degli Stati membri, i quali hanno l obbligo di recepirle
Servizio Accertamenti a Tutela della Fede Pubblica CCIAA Milano
 Pubblica CCIAA Milano La competenza Le funzioni di autorità di vigilanza per il controllo della conformità dei giocattoli alle disposizioni del presente decreto legislativo sono svolte dal Ministero dello
Pubblica CCIAA Milano La competenza Le funzioni di autorità di vigilanza per il controllo della conformità dei giocattoli alle disposizioni del presente decreto legislativo sono svolte dal Ministero dello
Il processo di marcatura CE riferito agli impianti gas medicali
 Il processo di marcatura CE riferito agli impianti gas medicali Soggetti coinvolti nel processo di Marcatura CE Fabbricante: la persona fisica o giuridica responsabile della progettazione, della fabbricazione,
Il processo di marcatura CE riferito agli impianti gas medicali Soggetti coinvolti nel processo di Marcatura CE Fabbricante: la persona fisica o giuridica responsabile della progettazione, della fabbricazione,
Autore: Flavio Banfi Organizzazione: ITALCERT S.r.l. Intervento: Valutatori per la Certificazione CE di prodotto 2
 Convegno: SEMINARIO AICQ SICEV SICEP 20 maggio 2011 Intervento: Valutatori per la Certificazione CE di prodotto Autore: Flavio Banfi Direttore Tecnico ITALCERT S.r.l. ITALCERT : Certificazione sistemi
Convegno: SEMINARIO AICQ SICEV SICEP 20 maggio 2011 Intervento: Valutatori per la Certificazione CE di prodotto Autore: Flavio Banfi Direttore Tecnico ITALCERT S.r.l. ITALCERT : Certificazione sistemi
GESTIONE DEL RISCHIO NEI DISPOSITIVI MEDICI: DALLA CLASSIFICAZIONE ALLA COMMERCIALIZZAZIONE
 1 GESTIONE DEL RISCHIO NEI DISPOSITIVI MEDICI: DALLA CLASSIFICAZIONE ALLA COMMERCIALIZZAZIONE Ing. Enrico Perfler Eudax s.r.l. Milano, 23 Gennaio 2014 Indice 2 Il concetto di rischio nei dispositivi medici
1 GESTIONE DEL RISCHIO NEI DISPOSITIVI MEDICI: DALLA CLASSIFICAZIONE ALLA COMMERCIALIZZAZIONE Ing. Enrico Perfler Eudax s.r.l. Milano, 23 Gennaio 2014 Indice 2 Il concetto di rischio nei dispositivi medici
Newsletter del MARZO 2013. In sintesi:
 Newsletter del MARZO 2013 In sintesi: Entrata in vigore del regolamento CE 453/2010 CE E CLP _ Sostanze Chimiche_ Entrata in vigore regolamento CE 305/2011 CPR_ Prodotti da Costruzione_ Entrata in vigore
Newsletter del MARZO 2013 In sintesi: Entrata in vigore del regolamento CE 453/2010 CE E CLP _ Sostanze Chimiche_ Entrata in vigore regolamento CE 305/2011 CPR_ Prodotti da Costruzione_ Entrata in vigore
Sistemi di certificazione e accreditamento
 Sistemi di certificazione e accreditamento Beniamino Cenci Goga L accreditamento riduce i rischi delle imprese e dei clienti poiché garantisce che gli organismi accreditati sono in grado di portare a termine
Sistemi di certificazione e accreditamento Beniamino Cenci Goga L accreditamento riduce i rischi delle imprese e dei clienti poiché garantisce che gli organismi accreditati sono in grado di portare a termine
REGOLAMENTO (CE) N. 304/2008 DELLA COMMISSIONE
 N. 304/2008 DELLA COMMISSIONE") I L 92/12 Gazzetta ufficiale dell Unione europea 3.4.2008 REGOLAMENO (CE) N. 304/2008 DELLA COMMISSIONE del 2 aprile 2008 che stabilisce, in conformità al regolamento (CE) n. 842/2006 del Parlamento europeo
I L 92/12 Gazzetta ufficiale dell Unione europea 3.4.2008 REGOLAMENO (CE) N. 304/2008 DELLA COMMISSIONE del 2 aprile 2008 che stabilisce, in conformità al regolamento (CE) n. 842/2006 del Parlamento europeo
Il processo di marcatura CE riferito agli impianti gas medicali
 Il processo di marcatura CE riferito agli impianti gas medicali P.I. Alessandro Melzi Funzione Valutazione Conformità Prodotto Laboratorio Dispositivi Medici IMQ S.p.A. - Milano POLITECNICO DI MILANO,
Il processo di marcatura CE riferito agli impianti gas medicali P.I. Alessandro Melzi Funzione Valutazione Conformità Prodotto Laboratorio Dispositivi Medici IMQ S.p.A. - Milano POLITECNICO DI MILANO,
Il processo di certificazione del software MD
 Il processo di certificazione del software MD Ing. Maurizio Bianchi Responsabile Tecnico Dispositivi Medici - IMQ S.p.A. maurizio.bianchi@imq.it Vicenza, 5 ottobre 2007 Introduzione Moduli certificativi
Il processo di certificazione del software MD Ing. Maurizio Bianchi Responsabile Tecnico Dispositivi Medici - IMQ S.p.A. maurizio.bianchi@imq.it Vicenza, 5 ottobre 2007 Introduzione Moduli certificativi
REGOLAMENTO PER LA DISCIPLINA
 C O M U N E D I B R U I N O PROVINCIA DI TORINO - C. A. P. 10090 REGOLAMENTO PER LA DISCIPLINA DELL ALBO PRETORIO DIGITALE Approvato con deliberazione della Giunta Comunale n. 34 del 14/4/2011 Depositato
C O M U N E D I B R U I N O PROVINCIA DI TORINO - C. A. P. 10090 REGOLAMENTO PER LA DISCIPLINA DELL ALBO PRETORIO DIGITALE Approvato con deliberazione della Giunta Comunale n. 34 del 14/4/2011 Depositato
La sorveglianza sui dispositivi medici
 Corso Attività di Polizia Sanitaria ROMA, 21 giugno 2012 La sorveglianza sui dispositivi medici Dr. Franco Abbenda Ministero della Salute DGFDSC Ufficio 03 1 Origini regolatorie - Armamentario del medico
Corso Attività di Polizia Sanitaria ROMA, 21 giugno 2012 La sorveglianza sui dispositivi medici Dr. Franco Abbenda Ministero della Salute DGFDSC Ufficio 03 1 Origini regolatorie - Armamentario del medico
UNI EN ISO 9001:2008 Sistemi di Gestione per la Qualità: requisiti e guida per l uso
 SORVEGLIANZA E CERTIFICAZIONI UNI EN ISO 9001:2008 Sistemi di Gestione per la Qualità: requisiti e guida per l uso Pagina 1 di 10 INTRODUZIONE La Norma UNI EN ISO 9001:2008 fa parte delle norme Internazionali
SORVEGLIANZA E CERTIFICAZIONI UNI EN ISO 9001:2008 Sistemi di Gestione per la Qualità: requisiti e guida per l uso Pagina 1 di 10 INTRODUZIONE La Norma UNI EN ISO 9001:2008 fa parte delle norme Internazionali
Centro Tecnico per la Rete Unitaria della Pubblica Amministrazione
 Centro Tecnico per la Rete Unitaria della Pubblica Amministrazione Area Rete Unitaria - Sezione Interoperabilità Linee guida del servizio di trasmissione di documenti informatici mediante posta elettronica
Centro Tecnico per la Rete Unitaria della Pubblica Amministrazione Area Rete Unitaria - Sezione Interoperabilità Linee guida del servizio di trasmissione di documenti informatici mediante posta elettronica
REPERTORIO AZIENDALE DEI DISPOSITIVI MEDICI
 S.O.C. Assistenza Farmaceutica AZIENDA N 4 Medio Friuli REPERTORIO DEI DISPOSITIVI MEDICI Edizione 0 del 20/11/07 REPERTORIO AZIENDALE DEI DISPOSITIVI MEDICI INDICE: 1 - Campo di applicazione 2 - Scopo
S.O.C. Assistenza Farmaceutica AZIENDA N 4 Medio Friuli REPERTORIO DEI DISPOSITIVI MEDICI Edizione 0 del 20/11/07 REPERTORIO AZIENDALE DEI DISPOSITIVI MEDICI INDICE: 1 - Campo di applicazione 2 - Scopo
Regolamento (CE) n. 1028/2006 del Consiglio del 19 giugno 2006 recante norme di commercializzazione applicabili alle uova.
 n. 1028/2006 del Consiglio del 19 giugno 2006 recante norme di commercializzazione applicabili alle uova.") Regolamento (CE) n. 1028/2006 del 19 giugno 2006. Regolamento (CE) n. 1028/2006 del Consiglio del 19 giugno 2006 recante norme di commercializzazione applicabili alle uova. (pubbl. in Gazz. Uff. dell Unione
Regolamento (CE) n. 1028/2006 del 19 giugno 2006. Regolamento (CE) n. 1028/2006 del Consiglio del 19 giugno 2006 recante norme di commercializzazione applicabili alle uova. (pubbl. in Gazz. Uff. dell Unione
Come cambiano gli obblighi per le strutture metalliche a partire dal 1 luglio 2014
 Regolamento (UE) n.305/11 e norme del gruppo EN1090 La Marcatura CE delle strutture metalliche alla luce delle Norme tecniche per le Costruzioni Firenze, 30/10/2014 Come cambiano gli obblighi per le strutture
Regolamento (UE) n.305/11 e norme del gruppo EN1090 La Marcatura CE delle strutture metalliche alla luce delle Norme tecniche per le Costruzioni Firenze, 30/10/2014 Come cambiano gli obblighi per le strutture
Marcatura CE. Controllo di produzione in fabbrica - FPC. Segnaletica stradale. Caratteristiche energetiche prodotti per edilizia
 ICMQ Certificazioni e controlli per le costruzioni La certificazione dei prodotti Le tipologie di certificazione rilasciate da ICMQ riguardano sia le certificazioni cogenti, cioè obbligatorie secondo la
ICMQ Certificazioni e controlli per le costruzioni La certificazione dei prodotti Le tipologie di certificazione rilasciate da ICMQ riguardano sia le certificazioni cogenti, cioè obbligatorie secondo la
Fascicolo tecnico e dichiarazione di conformità
 ISTITUTO DI RICERCHE E COLLAUDI M. MASINI S.r.l. Fascicolo tecnico e dichiarazione di conformità Camera Commercio di Padova 14 aprile 2011 Relatore: Gabriele Lualdi 173 Documenti a sostegno della Valutazione
ISTITUTO DI RICERCHE E COLLAUDI M. MASINI S.r.l. Fascicolo tecnico e dichiarazione di conformità Camera Commercio di Padova 14 aprile 2011 Relatore: Gabriele Lualdi 173 Documenti a sostegno della Valutazione
 Le regole del parlamento europeo per garantire qualità e sicurezza nel trattamento di cellule e tessuti umani (Direttiva 2004/23/CE) E stata recentemente recepita anche dall Italia, la direttiva europea
Le regole del parlamento europeo per garantire qualità e sicurezza nel trattamento di cellule e tessuti umani (Direttiva 2004/23/CE) E stata recentemente recepita anche dall Italia, la direttiva europea
Il glossario della Posta Elettronica Certificata (PEC) Diamo una definizione ai termini tecnici relativi al mondo della PEC.
 Diamo una definizione ai termini tecnici relativi al mondo della PEC.") Il glossario della Posta Elettronica Certificata (PEC) Diamo una definizione ai termini tecnici relativi al mondo della PEC. Avviso di mancata consegna L avviso, emesso dal sistema, per indicare l anomalia
Il glossario della Posta Elettronica Certificata (PEC) Diamo una definizione ai termini tecnici relativi al mondo della PEC. Avviso di mancata consegna L avviso, emesso dal sistema, per indicare l anomalia
Societa di Scienze Farmacologiche Applicate
 Societa di Scienze Farmacologiche Applicate Principali Processi Applicativi per lo Sviluppo e Commercializzazione di Dispositivi Medici - Seminario Gruppo di Lavoro Dispositivi Medici Auditorium Bayer
Societa di Scienze Farmacologiche Applicate Principali Processi Applicativi per lo Sviluppo e Commercializzazione di Dispositivi Medici - Seminario Gruppo di Lavoro Dispositivi Medici Auditorium Bayer
Direttiva Macchine 2006/42/CE. Dichiarazione CE di Conformità: la corretta compilazione.
 Direttiva Macchine 2006/42/CE Dichiarazione CE di Conformità: la corretta compilazione. Fonte: Guida Ufficiale Commissione europea Direttiva macchine 2006/42/CE Ed. 2010 Dati fabbricante La ragione sociale
Direttiva Macchine 2006/42/CE Dichiarazione CE di Conformità: la corretta compilazione. Fonte: Guida Ufficiale Commissione europea Direttiva macchine 2006/42/CE Ed. 2010 Dati fabbricante La ragione sociale
della manutenzione, includa i requisiti relativi ai sottosistemi strutturali all interno del loro contesto operativo.
 L 320/8 Gazzetta ufficiale dell Unione europea IT 17.11.2012 REGOLAMENTO (UE) N. 1078/2012 DELLA COMMISSIONE del 16 novembre 2012 relativo a un metodo di sicurezza comune per il monitoraggio che devono
L 320/8 Gazzetta ufficiale dell Unione europea IT 17.11.2012 REGOLAMENTO (UE) N. 1078/2012 DELLA COMMISSIONE del 16 novembre 2012 relativo a un metodo di sicurezza comune per il monitoraggio che devono
Qualità e Certificazione certificazione di prodotto - Marcantonio Catelani Facoltà di Ingegneria Firenze
 Qualità e Certificazione certificazione di prodotto - Marcantonio Catelani Facoltà di Ingegneria Firenze 1 Focus sulla Certificazione Organismi di certificazione Certificazione personale Certificazione
Qualità e Certificazione certificazione di prodotto - Marcantonio Catelani Facoltà di Ingegneria Firenze 1 Focus sulla Certificazione Organismi di certificazione Certificazione personale Certificazione
PIANO DI CONSERVAZIONE DEI DOCUMENTI
 PIANO DI CONSERVAZIONE DEI DOCUMENTI Documento n. 8 - Allegato al manuale di gestione PIANO DI CONSERVAZIONE DEI DOCUMENTI 1. Composizione del piano Il piano di conservazione oltre che dai seguenti articoli
PIANO DI CONSERVAZIONE DEI DOCUMENTI Documento n. 8 - Allegato al manuale di gestione PIANO DI CONSERVAZIONE DEI DOCUMENTI 1. Composizione del piano Il piano di conservazione oltre che dai seguenti articoli
Additivi alimentari: aspetti normativi e criticità della etichettatura
 Corso di formazione sugli additivi alimentari 23 gennaio 2013 I edizione 24 gennaio 2013 II edizione Benevento Additivi alimentari: aspetti normativi e criticità della etichettatura Antonella Semeraro
Corso di formazione sugli additivi alimentari 23 gennaio 2013 I edizione 24 gennaio 2013 II edizione Benevento Additivi alimentari: aspetti normativi e criticità della etichettatura Antonella Semeraro
(Atti non legislativi) DECISIONI
 DECISIONI") 24.2.2015 Gazzetta ufficiale dell Unione europea L 52/1 II (Atti non legislativi) DECISIONI DECISIONE DI ESECUZIONE (UE) 2015/261 DELLA COMMISSIONE del 6 febbraio 2015 che modifica le decisioni 2010/470/UE
24.2.2015 Gazzetta ufficiale dell Unione europea L 52/1 II (Atti non legislativi) DECISIONI DECISIONE DI ESECUZIONE (UE) 2015/261 DELLA COMMISSIONE del 6 febbraio 2015 che modifica le decisioni 2010/470/UE
LINEE GUIDA IN MERITO ALL ACQUISTO E ALL USO DI MACCHINE E/O APPARECCHI ELETTRICI SOMMARIO
 Pag. 1 / 6 1 MODIFICHE 2 PREMESSA 3 DEFINIZIONI SOMMARIO 4 PRINCPALI NORMATIVE DI RIFERIMENTO 5 MESSA IN SERVIZIO E UTILIZZO 6 INDICAZIONI PER UN CORRETTO ACQUISTO 7 SICUREZZA E MARCATURA CE 8 PROTOTIPI
Pag. 1 / 6 1 MODIFICHE 2 PREMESSA 3 DEFINIZIONI SOMMARIO 4 PRINCPALI NORMATIVE DI RIFERIMENTO 5 MESSA IN SERVIZIO E UTILIZZO 6 INDICAZIONI PER UN CORRETTO ACQUISTO 7 SICUREZZA E MARCATURA CE 8 PROTOTIPI
Sessione Monitoraggio dei consumi dei dispositivi medici: strumenti di analisi per valutazioni economiche e di performance IL CONTRIBUTO DELLE REGIONI
 Sessione Monitoraggio dei consumi dei dispositivi medici: strumenti di analisi per valutazioni economiche e di performance IL CONTRIBUTO DELLE REGIONI Dott. Loredano Giorni Direzione Generale diritti di
Sessione Monitoraggio dei consumi dei dispositivi medici: strumenti di analisi per valutazioni economiche e di performance IL CONTRIBUTO DELLE REGIONI Dott. Loredano Giorni Direzione Generale diritti di
Pubblicazione delle nuove Direttive Comunitarie
 Pubblicazione delle nuove Direttive Comunitarie Milano, 21 Ottobre 2014 Ivan Furcas ivan.furcas@it.bureauveritas.com Nuovo quadro legislativo Pubblicazione L96 GUCE (Gazzetta Ufficiale Comunità Europea)
Pubblicazione delle nuove Direttive Comunitarie Milano, 21 Ottobre 2014 Ivan Furcas ivan.furcas@it.bureauveritas.com Nuovo quadro legislativo Pubblicazione L96 GUCE (Gazzetta Ufficiale Comunità Europea)
La norma ISO 9001:08 ha apportato modifiche alla normativa precedente in
 La norma ISO 9001:08 ha apportato modifiche alla normativa precedente in base alle necessità di chiarezza emerse nell utilizzo della precedente versione e per meglio armonizzarla con la ISO 14001:04. Elemento
La norma ISO 9001:08 ha apportato modifiche alla normativa precedente in base alle necessità di chiarezza emerse nell utilizzo della precedente versione e per meglio armonizzarla con la ISO 14001:04. Elemento
MANUALE DELLA QUALITA Revisione: Sezione 4 SISTEMA DI GESTIONE PER LA QUALITA
 Pagina: 1 di 5 SISTEMA DI GESTIONE PER LA QUALITA 4.0 SCOPO DELLA SEZIONE Illustrare la struttura del Sistema di Gestione Qualità SGQ dell Istituto. Per gli aspetti di dettaglio, la Procedura di riferimento
Pagina: 1 di 5 SISTEMA DI GESTIONE PER LA QUALITA 4.0 SCOPO DELLA SEZIONE Illustrare la struttura del Sistema di Gestione Qualità SGQ dell Istituto. Per gli aspetti di dettaglio, la Procedura di riferimento
LA CERTIFICAZIONE. Dr.ssa Eletta Cavedoni Responsabile Qualità Cosmolab srl Tortona
 LA CERTIFICAZIONE Dr.ssa Eletta Cavedoni Responsabile Qualità Cosmolab srl Tortona Qualità Grado in cui un insieme di caratteristiche intrinseche soddisfa i requisiti (UNI EN ISO 9000/00) Requisito Esigenza
LA CERTIFICAZIONE Dr.ssa Eletta Cavedoni Responsabile Qualità Cosmolab srl Tortona Qualità Grado in cui un insieme di caratteristiche intrinseche soddisfa i requisiti (UNI EN ISO 9000/00) Requisito Esigenza
Delibera n. 129/04 L AUTORITÀ PER L ENERGIA ELETTRICA E IL GAS. Nella riunione del 22 luglio 2004. Visti:
 Pagina 1 di 5 Pubblicata sul sito www.autorita.energia.it il 29 luglio 2004, ai sensi dell'articolo 6, comma 4, della deliberazione dell'autorità per l'energia elettrica e il gas 20 febbraio 2001, n. 26/01
Pagina 1 di 5 Pubblicata sul sito www.autorita.energia.it il 29 luglio 2004, ai sensi dell'articolo 6, comma 4, della deliberazione dell'autorità per l'energia elettrica e il gas 20 febbraio 2001, n. 26/01
REGOLAMENTO (CE) N. 183/2005 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO
 N. 183/2005 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO") REGOLAMENTO (CE) N. 183/2005 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO del 12 gennaio 2005 che stabilisce requisiti per l igiene dei mangimi Regolamento 183/2005 Applicazione sul territorio regionale L applicazione
REGOLAMENTO (CE) N. 183/2005 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO del 12 gennaio 2005 che stabilisce requisiti per l igiene dei mangimi Regolamento 183/2005 Applicazione sul territorio regionale L applicazione
4. RISORSE STRUTTURALI E TECNOLOGICHE
 136 4. RISORSE STRUTTURALI E TECNOLOGICHE 4.5 ELENCO DOCUMENTAZIONE 137 ELENCO DELLA DOCUMENTAZIONE E MATRICE DELLE RESPONSABILITÀ AC 190903 00 SGRS1.1A040 a 01 atto formale con il quale viene identificato
136 4. RISORSE STRUTTURALI E TECNOLOGICHE 4.5 ELENCO DOCUMENTAZIONE 137 ELENCO DELLA DOCUMENTAZIONE E MATRICE DELLE RESPONSABILITÀ AC 190903 00 SGRS1.1A040 a 01 atto formale con il quale viene identificato
PRESCRIZIONI PARTICOLARI DIRETTIVA 2006/42/CE RELATIVA ALLE MACCHINE Allegato X Garanzia Qualità Totale
 Titolo PRESCRIZIONI PARTICOLARI DIRETTIVA 2006/42/CE RELATIVA ALLE MACCHINE Allegato X Garanzia Qualità Totale Riferimento Data entrata in vigore Approvato da PR PART ON/MACC/X Rev. 0 del 01/06/2016 IMQ
Titolo PRESCRIZIONI PARTICOLARI DIRETTIVA 2006/42/CE RELATIVA ALLE MACCHINE Allegato X Garanzia Qualità Totale Riferimento Data entrata in vigore Approvato da PR PART ON/MACC/X Rev. 0 del 01/06/2016 IMQ
Le direttive nuovo approccio applicabili alle macchine in riferimento alla direttiva 2006/42/CE
 Le direttive nuovo approccio applicabili alle macchine in riferimento alla direttiva 2006/42/CE Cuneo, 8 Ottobre 2013 Ivan Furcas ivan.furcas@it.bureauveritas.com Sviluppo della legislazione di prodotto
Le direttive nuovo approccio applicabili alle macchine in riferimento alla direttiva 2006/42/CE Cuneo, 8 Ottobre 2013 Ivan Furcas ivan.furcas@it.bureauveritas.com Sviluppo della legislazione di prodotto
OBBLIGHI IN MATERIA DI SICUREZZA PER DATORI DI LAVORO, PREPOSTI, DIRIGENTI E LAVORATORI
 OBBLIGHI IN MATERIA DI SICUREZZA PER DATORI DI LAVORO, PREPOSTI, DIRIGENTI E LAVORATORI Gli accordi Stato- Regioni del 21 dicembre sono stati pubblicati sulla Gazzetta Ufficiale n. 8 dell 11 gennaio 2012.
OBBLIGHI IN MATERIA DI SICUREZZA PER DATORI DI LAVORO, PREPOSTI, DIRIGENTI E LAVORATORI Gli accordi Stato- Regioni del 21 dicembre sono stati pubblicati sulla Gazzetta Ufficiale n. 8 dell 11 gennaio 2012.
Sistema europeo di regolamentazione per i dispositivi medici
 Sistema europeo di regolamentazione per i dispositivi medici Il campo di gioco Nuovo Approccio all'armonizzazione tecnica e Approccio Globale alla valutazione della conformità L'articolo 95 paragrafo 3
Sistema europeo di regolamentazione per i dispositivi medici Il campo di gioco Nuovo Approccio all'armonizzazione tecnica e Approccio Globale alla valutazione della conformità L'articolo 95 paragrafo 3
visto il trattato che istituisce la Comunità europea, in particolare l articolo 93, vista la proposta della Commissione,
 IL CONSIGLIO DELL UNIONE EUROPEA, visto il trattato che istituisce la Comunità europea, in particolare l articolo 93, vista la proposta della Commissione, (2) Per assicurare la corretta applicazione dell
IL CONSIGLIO DELL UNIONE EUROPEA, visto il trattato che istituisce la Comunità europea, in particolare l articolo 93, vista la proposta della Commissione, (2) Per assicurare la corretta applicazione dell
IL MINISTRO DEL LAVORO E DELLE POLITICHE SOCIALI di concerto con IL MINISTRO DELLA SALUTE
 Decreto del Ministero dell interno 4 febbraio 2011 Definizione dei criteri per il rilascio delle autorizzazioni di cui all art. 82, comma 2, del D.Lgs. 09/04/2008, n. 81, e successive modifiche ed integrazioni.
Decreto del Ministero dell interno 4 febbraio 2011 Definizione dei criteri per il rilascio delle autorizzazioni di cui all art. 82, comma 2, del D.Lgs. 09/04/2008, n. 81, e successive modifiche ed integrazioni.
Valutazione dei Rischi. Normativa e documenti di riferimento. Definizioni (UNI EN ISO 12100)
") Definizione e individuazione dei fattori di rischio, individuazione delle misure di prevenzione e protezione, riunione periodica Rev. 1 del 28/11/2012 ARISSA Maggio 2015 2 Normativa e documenti di riferimento
Definizione e individuazione dei fattori di rischio, individuazione delle misure di prevenzione e protezione, riunione periodica Rev. 1 del 28/11/2012 ARISSA Maggio 2015 2 Normativa e documenti di riferimento
CITTÀ DI AGROPOLI. Regolamento per la pubblicazione delle Determinazioni sul sito internet istituzionale dell Ente
 CITTÀ DI AGROPOLI Regolamento per la pubblicazione delle Determinazioni sul sito internet istituzionale dell Ente Approvato con deliberazione della Giunta comunale n 358 del 06.12.2012 Regolamento per
CITTÀ DI AGROPOLI Regolamento per la pubblicazione delle Determinazioni sul sito internet istituzionale dell Ente Approvato con deliberazione della Giunta comunale n 358 del 06.12.2012 Regolamento per
Decreto Legislativo 49/2014. Moduli Fotovoltaici e RAEE
 Decreto Legislativo 49/2014 Moduli Fotovoltaici e RAEE 1 Indice Introduzione... 3 Q1 La mia società rientra nella definizione di produttore di AEE (Apparecchiature Elettriche ed Elettroniche)?... 4 Q2
Decreto Legislativo 49/2014 Moduli Fotovoltaici e RAEE 1 Indice Introduzione... 3 Q1 La mia società rientra nella definizione di produttore di AEE (Apparecchiature Elettriche ed Elettroniche)?... 4 Q2
LE NORME E LA CERTIFICAZIONE
 LE NORME E LA CERTIFICAZIONE Introduzione alla Qualità 1 DEFINIZIONE DI NORMA Documento, prodotto mediante consenso ed approvato da un organismo riconosciuto, che fornisce, per usi comuni e ripetuti, regole,
LE NORME E LA CERTIFICAZIONE Introduzione alla Qualità 1 DEFINIZIONE DI NORMA Documento, prodotto mediante consenso ed approvato da un organismo riconosciuto, che fornisce, per usi comuni e ripetuti, regole,
La sorveglianza del mercato alla luce del nuovo regolamento 528/2012 (UE)
") 1 La sorveglianza del mercato alla luce del nuovo regolamento 528/2012 (UE) M A R I S T E L L A R U B B I A N I C S C - ISS ROMA Monitoraggio delle Conformità e Sorveglianza del Mercato Attività di controllo
1 La sorveglianza del mercato alla luce del nuovo regolamento 528/2012 (UE) M A R I S T E L L A R U B B I A N I C S C - ISS ROMA Monitoraggio delle Conformità e Sorveglianza del Mercato Attività di controllo
Attestato di prestazione energetica per edificio residenziale
 Attestato di prestazione energetica per edificio residenziale Ubicazione Proprietà Tipologia edilizia Riferimenti catastali Codice attestato San Casciano Val Di Pesa, via Decimo 14, 50026 San Casciano
Attestato di prestazione energetica per edificio residenziale Ubicazione Proprietà Tipologia edilizia Riferimenti catastali Codice attestato San Casciano Val Di Pesa, via Decimo 14, 50026 San Casciano
Agenti chimici: il regolamento CLP
 Informazioni sulla tutela della salute e della sicurezza dei lavoratori Gennaio 2013 Pillole di sicurezza A cura del RSPP e dell Unità Organizzativa a Supporto del Servizio di Prevenzione e Protezione
Informazioni sulla tutela della salute e della sicurezza dei lavoratori Gennaio 2013 Pillole di sicurezza A cura del RSPP e dell Unità Organizzativa a Supporto del Servizio di Prevenzione e Protezione
Il Ministro delle politiche agricole alimentari e forestali
 Misure urgenti per il miglioramento del sistema di controllo come disciplinato agli artt. 27 e seguenti del Reg. (CE) n. 834/2007 e relativi regolamenti di applicazione. VISTO il Reg. (CE) n. 834/2007
Misure urgenti per il miglioramento del sistema di controllo come disciplinato agli artt. 27 e seguenti del Reg. (CE) n. 834/2007 e relativi regolamenti di applicazione. VISTO il Reg. (CE) n. 834/2007
DECRETO 14 febbraio 1997 ( Gazz. Uff. n. 58 11 marzo 1997 )
") DECRETO 14 febbraio 1997 ( Gazz. Uff. n. 58 11 marzo 1997 ) Determinazione del tipo, modalità e periodicità del controllo di qualità da parte del fisico specialista o dell esperto qualificato delle apparecchiature
DECRETO 14 febbraio 1997 ( Gazz. Uff. n. 58 11 marzo 1997 ) Determinazione del tipo, modalità e periodicità del controllo di qualità da parte del fisico specialista o dell esperto qualificato delle apparecchiature
Direttiva Macchine e Sicurezza in Cantiere
 Ordine degli Ingegneri della Provincia di Palermo con il patrocinio di AICQ Sicilia Direttiva Macchine e Sicurezza in Cantiere Palermo, 8-11-2013 c/o Ordine Ingegneri Palermo IL PROCESSO DI RIDUZIONE DEI
Ordine degli Ingegneri della Provincia di Palermo con il patrocinio di AICQ Sicilia Direttiva Macchine e Sicurezza in Cantiere Palermo, 8-11-2013 c/o Ordine Ingegneri Palermo IL PROCESSO DI RIDUZIONE DEI
GMP e norme ISO nella produzione dei dispositivi medici. Antonella Mamoli Roma, 19 Maggio 2009
 GMP e norme ISO nella produzione dei dispositivi medici Antonella Mamoli Roma, 19 Maggio 2009 GMP: riferimento normativo per i produttori farmaceutici L attuale normativa italiana del farmaco Il Decreto
GMP e norme ISO nella produzione dei dispositivi medici Antonella Mamoli Roma, 19 Maggio 2009 GMP: riferimento normativo per i produttori farmaceutici L attuale normativa italiana del farmaco Il Decreto
Marcatura CE Cancelli
 SB studio www.essebistudio.com www.marcatura-ce-cancelli.com Marcatura CE Cancelli SB studio di Bertolo Sergio Borgo Treviso 152/C 31033 Castelfranco Veneto TV Direttiva Prodotti da Costruzione CPD 89/106/CEE
SB studio www.essebistudio.com www.marcatura-ce-cancelli.com Marcatura CE Cancelli SB studio di Bertolo Sergio Borgo Treviso 152/C 31033 Castelfranco Veneto TV Direttiva Prodotti da Costruzione CPD 89/106/CEE
REGIONE SICILIANA Universitaria Vittorio Emanuele, Ferrarotto, S. Bambino Catania ACCREDITAMENTO ISTITUZIONALE - I PARTE -
 110 4. RISORSE STRUTTURALI E TECNOLOGICHE 4.3 111 Le attività da svolgere per soddisfare i requisiti relativi alle risorse umane e tecnologiche consistono nella: 4.3.1 identificazione di un referente per
110 4. RISORSE STRUTTURALI E TECNOLOGICHE 4.3 111 Le attività da svolgere per soddisfare i requisiti relativi alle risorse umane e tecnologiche consistono nella: 4.3.1 identificazione di un referente per
Direttiva Macchine2006/42/CE
 PAG. 1 DI 5 REV. 00 SAVE DATA: 09/10/12 PRINT DATA: 10/10/12 Direttiva Macchine2006/42/CE Definizione di immissione sul mercato Indicazioni tratte da Guida all applicazione della direttiva macchine 2006/42/CE
PAG. 1 DI 5 REV. 00 SAVE DATA: 09/10/12 PRINT DATA: 10/10/12 Direttiva Macchine2006/42/CE Definizione di immissione sul mercato Indicazioni tratte da Guida all applicazione della direttiva macchine 2006/42/CE
MANUALE DELLA QUALITÀ Pag. 1 di 6
 MANUALE DELLA QUALITÀ Pag. 1 di 6 INDICE GESTIONE DELLE RISORSE Messa a disposizione delle risorse Competenza, consapevolezza, addestramento Infrastrutture Ambiente di lavoro MANUALE DELLA QUALITÀ Pag.
MANUALE DELLA QUALITÀ Pag. 1 di 6 INDICE GESTIONE DELLE RISORSE Messa a disposizione delle risorse Competenza, consapevolezza, addestramento Infrastrutture Ambiente di lavoro MANUALE DELLA QUALITÀ Pag.
Il Riconoscimento della formazione pregressa e gli organismi paritetici
 Ing. Marco CONTI SEMINARIO FORMATIVO Il Riconoscimento della formazione pregressa e gli organismi paritetici Il riconoscimento della formazione pregressa per lavoratori e datori di lavoro RICONOSCIMENTO
Ing. Marco CONTI SEMINARIO FORMATIVO Il Riconoscimento della formazione pregressa e gli organismi paritetici Il riconoscimento della formazione pregressa per lavoratori e datori di lavoro RICONOSCIMENTO
cenni sulle direttive macchine e di prodotto
 9(1(72 cenni sulle direttive macchine e di prodotto 5HODWRUH,QJ5REHUWR5LQDOGL /HGLUHWWLYHFRPXQLWDULHVFRSRH DSSOLFD]LRQH /H'LUHWWLYH&RPXQLWDULHVRQRODSULQFLSDOHIRQWHGHOGLULWWRGD FXL GHULYDODOHJLVOD]LRQHFKHKDVRVWLWXLWRHVRVWLWXLUjLQGHWHU
9(1(72 cenni sulle direttive macchine e di prodotto 5HODWRUH,QJ5REHUWR5LQDOGL /HGLUHWWLYHFRPXQLWDULHVFRSRH DSSOLFD]LRQH /H'LUHWWLYH&RPXQLWDULHVRQRODSULQFLSDOHIRQWHGHOGLULWWRGD FXL GHULYDODOHJLVOD]LRQHFKHKDVRVWLWXLWRHVRVWLWXLUjLQGHWHU
REGOLAMENTO SUL TRATTAMENTO DEI DATI PERSONALI
 COMUNE DI VIANO PROVINCIA DI REGGIO EMILIA REGOLAMENTO SUL TRATTAMENTO DEI DATI PERSONALI Approvato con deliberazione di G.C. n. 73 del 28.11.2000 INDICE TITOLO 1 ART. 1 ART. 2 ART. 3 ART. 4 ART. 5 ART.
COMUNE DI VIANO PROVINCIA DI REGGIO EMILIA REGOLAMENTO SUL TRATTAMENTO DEI DATI PERSONALI Approvato con deliberazione di G.C. n. 73 del 28.11.2000 INDICE TITOLO 1 ART. 1 ART. 2 ART. 3 ART. 4 ART. 5 ART.
E. Monica Russo. Integratori alimentari e novel food
 E. Monica Russo Integratori alimentari e novel food enza.russo@lab-to.camcom.it Isernia, 2 aprile 2014 1 MINISTERO DELLA SALUTE www.salute.gov.it TEMI E PROFESSIONI ALIMENTI ALIMENTI PARTICOLARI integratori
E. Monica Russo Integratori alimentari e novel food enza.russo@lab-to.camcom.it Isernia, 2 aprile 2014 1 MINISTERO DELLA SALUTE www.salute.gov.it TEMI E PROFESSIONI ALIMENTI ALIMENTI PARTICOLARI integratori
SCELTA DEL PERCORSO PER LA CERTIFICAZIONE DI UN DISPOSITIVO MEDICO
 51 SIMPOSIO AFI Dispositivi Medici: evoluzione dell ampia gamma dei borderline Rimini, 8 giugno 2011 SCELTA DEL PERCORSO PER LA CERTIFICAZIONE DI UN DISPOSITIVO MEDICO Antonella Mamoli - Fabio Geremia
51 SIMPOSIO AFI Dispositivi Medici: evoluzione dell ampia gamma dei borderline Rimini, 8 giugno 2011 SCELTA DEL PERCORSO PER LA CERTIFICAZIONE DI UN DISPOSITIVO MEDICO Antonella Mamoli - Fabio Geremia
Gli accordi definiscono la durata, i contenuti e le modalità della formazione da svolgere.
 Torino, 24 gennaio 2012 Oggetto: Accordo Stato Regioni per la formazione dei lavoratori ai sensi dell articolo 37, comma 2, del decreto legislativo 9 aprile 2008, n*81 NOTA INFORMATIVA Dopo quasi tre anni
Torino, 24 gennaio 2012 Oggetto: Accordo Stato Regioni per la formazione dei lavoratori ai sensi dell articolo 37, comma 2, del decreto legislativo 9 aprile 2008, n*81 NOTA INFORMATIVA Dopo quasi tre anni
MINISTERO DELLE INFRASTRUTTURE E DEI TRASPORTI
 MINISTERO DELLE INFRASTRUTTURE E DEI TRASPORTI DIPARTIMENTO per i TRASPORTI, la NAVIGAZIONE ed i SISTEMI INFORMATIVI e STATISTICI DIREZIONE GENERALE TERRITORIALE DEL NORD-EST CENTRO PROVA AUTOVEICOLI di
MINISTERO DELLE INFRASTRUTTURE E DEI TRASPORTI DIPARTIMENTO per i TRASPORTI, la NAVIGAZIONE ed i SISTEMI INFORMATIVI e STATISTICI DIREZIONE GENERALE TERRITORIALE DEL NORD-EST CENTRO PROVA AUTOVEICOLI di
La direttiva 98/79/CE: applicazione e classificazione dei dispositivi per diagnostica in vitro
 La direttiva 98/79/CE: applicazione e classificazione dei dispositivi per diagnostica in vitro European Diagnostic Manufacturers Association Natale Bova QA Manager Instrumentation Laboratory EDMA Regulatory
La direttiva 98/79/CE: applicazione e classificazione dei dispositivi per diagnostica in vitro European Diagnostic Manufacturers Association Natale Bova QA Manager Instrumentation Laboratory EDMA Regulatory
La banca dati EUDAMED nella nuova direttiva e i rapporti con il RDM
 La banca dati EUDAMED nella nuova direttiva e i rapporti con il RDM Claudia Biffoli Ministero della Salute Direzione Generale del Sistema Informativo II Conferenza Nazionale sui Dispositivi medici Sessione
La banca dati EUDAMED nella nuova direttiva e i rapporti con il RDM Claudia Biffoli Ministero della Salute Direzione Generale del Sistema Informativo II Conferenza Nazionale sui Dispositivi medici Sessione
(Testo rilevante ai fini del SEE) 9.8.2007 7.7.2010 9.10.1999. EN 12322:1999/A1:2001 31.7.2002 Nota 3 Data scaduta (30.4.2002) 19.8.2011 19.8.
 9.8.2007 7.7.2010 9.10.1999. EN 12322:1999/A1:2001 31.7.2002 Nota 3 Data scaduta (30.4.2002) 19.8.2011 19.8.") C 22/30 Gazzetta ufficiale dell Unione europea 24.1.2013 Comunicazione della Commissione nell ambito dell applicazione della direttiva 98/79/CE del Parlamento europeo e del Consiglio, del 27 ottobre 1998,
C 22/30 Gazzetta ufficiale dell Unione europea 24.1.2013 Comunicazione della Commissione nell ambito dell applicazione della direttiva 98/79/CE del Parlamento europeo e del Consiglio, del 27 ottobre 1998,
FEDERATION EUROPEENNE DE LA MANUTENTION Gruppo di prodotto Piattaforme di lavoro mobili elevabili
 FEDERATION EUROPEENNE DE LA MANUTENTION Gruppo di prodotto Piattaforme di lavoro mobili elevabili FEM Breve guida all identificazione delle piattaforme di lavoro mobili elevabili non conformi 05.2012 (I)
FEDERATION EUROPEENNE DE LA MANUTENTION Gruppo di prodotto Piattaforme di lavoro mobili elevabili FEM Breve guida all identificazione delle piattaforme di lavoro mobili elevabili non conformi 05.2012 (I)
DEFINIZIONI INDISPENSABILI
 1 DEFINIZIONI INDISPENSABILI Preimballaggio Per imballaggio preconfezionato, o preimballaggio, si intende l insieme del prodotto e dell imballaggio nel quale è confezionato. Possiamo affermare quindi che
1 DEFINIZIONI INDISPENSABILI Preimballaggio Per imballaggio preconfezionato, o preimballaggio, si intende l insieme del prodotto e dell imballaggio nel quale è confezionato. Possiamo affermare quindi che
PROCEDURA DI GESTIONE DELLE PRESCRIZIONI LEGALI
 legali 0 18/05/09 1 6 PROCEDURA DI GESTIONE DELLE PRESCRIZIONI LEGALI INDICE PROCEDURA DI GESTIONE DELLE PRESCRIZIONI LEGALI...1 INDICE...1 1 Scopo...2 2 Campo di applicazione...2 3 Terminologia ed abbreviazioni...2
legali 0 18/05/09 1 6 PROCEDURA DI GESTIONE DELLE PRESCRIZIONI LEGALI INDICE PROCEDURA DI GESTIONE DELLE PRESCRIZIONI LEGALI...1 INDICE...1 1 Scopo...2 2 Campo di applicazione...2 3 Terminologia ed abbreviazioni...2
REGOLAMENTO PER LA PUBBLICAZIONE DI ATTI E PROVVEDIMENTI ALL ALBO CAMERALE. (Adottato con delibera della Giunta Camerale n.72, del 17 ottobre 2014)
") REGOLAMENTO PER LA PUBBLICAZIONE DI ATTI E PROVVEDIMENTI ALL ALBO CAMERALE. (Adottato con delibera della Giunta Camerale n.72, del 17 ottobre 2014) Art.1 - Oggetto Il presente Regolamento disciplina, ai
REGOLAMENTO PER LA PUBBLICAZIONE DI ATTI E PROVVEDIMENTI ALL ALBO CAMERALE. (Adottato con delibera della Giunta Camerale n.72, del 17 ottobre 2014) Art.1 - Oggetto Il presente Regolamento disciplina, ai
REGOLAMENTO PER LA GESTIONE DELLE PROCEDURE DI PUBBLICAZIONE ALL ALBO PRETORIO ONLINE
 REGOLAMENTO PER LA GESTIONE DELLE PROCEDURE DI PUBBLICAZIONE ALL ALBO PRETORIO ONLINE (appendice al regolamento sull ordinamento degli uffici e dei servizi) Approvato con delibera di G.C. n. 6 del 27.01.2011
REGOLAMENTO PER LA GESTIONE DELLE PROCEDURE DI PUBBLICAZIONE ALL ALBO PRETORIO ONLINE (appendice al regolamento sull ordinamento degli uffici e dei servizi) Approvato con delibera di G.C. n. 6 del 27.01.2011
PROCEDURE STANDARDIZZATE PER LA VALUTAZIONE DEI RISCHI DELLE PMI
 N.30 PDF Numero 10 PDF - anno 2012 DIRETTORE RINO PAVANELLO Rivista Ambiente e Lavoro 2012 PDF Manuale Tecnico-giuridico di In-formazione e Documentazione per RSPP, RLS, Giuristi, Operatori, Tecnici e
N.30 PDF Numero 10 PDF - anno 2012 DIRETTORE RINO PAVANELLO Rivista Ambiente e Lavoro 2012 PDF Manuale Tecnico-giuridico di In-formazione e Documentazione per RSPP, RLS, Giuristi, Operatori, Tecnici e
ISTITUTO SUPERIORE PER LA PREVENZIONE E LA SICUREZZA DEL LAVORO Dipartimento di Roma
 ISTITUTO SUPERIORE PER LA PREVENZIONE E LA SICUREZZA DEL LAVORO Dipartimento di Roma CORSO di FORMAZIONE per RSPP e ASPP Modulo A D.LGS. 81/08 I dispositivi di protezione individuale (Ing.. Ugo Romano)
ISTITUTO SUPERIORE PER LA PREVENZIONE E LA SICUREZZA DEL LAVORO Dipartimento di Roma CORSO di FORMAZIONE per RSPP e ASPP Modulo A D.LGS. 81/08 I dispositivi di protezione individuale (Ing.. Ugo Romano)
Organizzazioni di volontariato: quadro sintetico degli adempimenti in materia di salute e sicurezza dei lavoratori.
 Sede operativa via Ricasoli, 9-50122 Firenze Sede Legale via de' Martelli 8-50129 Firenze Tel. 055 271731 - Fax 055 214720 http://www.cesvot.it Organizzazioni di volontariato: quadro sintetico degli adempimenti
Sede operativa via Ricasoli, 9-50122 Firenze Sede Legale via de' Martelli 8-50129 Firenze Tel. 055 271731 - Fax 055 214720 http://www.cesvot.it Organizzazioni di volontariato: quadro sintetico degli adempimenti
CONVEGNO regionale DIRIGENTI SCOLASTICI LA GESTIONE DELLA SICUREZZA NELLE ISTTITUZIONI SCOLASTICHE: 1 Dicembre 2014
 CONVEGNO regionale DIRIGENTI SCOLASTICI LA GESTIONE DELLA SICUREZZA NELLE ISTTITUZIONI SCOLASTICHE: RUOLI E RESPONSABILITÀ 1 Dicembre 2014 IS E. MOLINARI Via Crescenzago, 110 - Milano Formazione ed informazione
CONVEGNO regionale DIRIGENTI SCOLASTICI LA GESTIONE DELLA SICUREZZA NELLE ISTTITUZIONI SCOLASTICHE: RUOLI E RESPONSABILITÀ 1 Dicembre 2014 IS E. MOLINARI Via Crescenzago, 110 - Milano Formazione ed informazione
Nuovo approccio all attività di sorveglianza sanitaria in Azienda. Capo III Sezione I, Art. 25 e Sezione V, Artt. 38 42 D.Lgs. 81/08 e s.m.i.
 Nuovo approccio all attività di sorveglianza sanitaria in Azienda Capo III Sezione I, Art. 25 e Sezione V, Artt. 38 42 D.Lgs. 81/08 e s.m.i. IL MEDICO COMPETENTE Collabora con il datore di lavoro e con
Nuovo approccio all attività di sorveglianza sanitaria in Azienda Capo III Sezione I, Art. 25 e Sezione V, Artt. 38 42 D.Lgs. 81/08 e s.m.i. IL MEDICO COMPETENTE Collabora con il datore di lavoro e con
COMUNE DI CASTELLAR (Provincia di Cuneo) PROGRAMMA TRIENNALE PER LA TRASPARENZA E L INTEGRITA TRIENNIO 2014/2016.
 PROGRAMMA TRIENNALE PER LA TRASPARENZA E L INTEGRITA TRIENNIO 2014/2016.") COMUNE DI CASTELLAR (Provincia di Cuneo) PROGRAMMA TRIENNALE PER LA TRASPARENZA E L INTEGRITA TRIENNIO 2014/2016. Indice: Premessa 1. FONTI NORMATIVE 2. STRUMENTI 3. DATI DA PUBBLICARE 4. INIZIATIVE DI
COMUNE DI CASTELLAR (Provincia di Cuneo) PROGRAMMA TRIENNALE PER LA TRASPARENZA E L INTEGRITA TRIENNIO 2014/2016. Indice: Premessa 1. FONTI NORMATIVE 2. STRUMENTI 3. DATI DA PUBBLICARE 4. INIZIATIVE DI
aggiorna le disposizioni per gli accertamenti documentali sugli impianti di utenza NUOVI (di nuova installazione);
;") FEBBRAIO 2014 Il 6 febbraio 2014 l Autorità per l Energia Elettrica e il Gas (AEEG) ha emanato e pubblicato sul proprio sito la Delibera n. 40/2014/R/gas, Disposizioni in materia di accertamenti della
FEBBRAIO 2014 Il 6 febbraio 2014 l Autorità per l Energia Elettrica e il Gas (AEEG) ha emanato e pubblicato sul proprio sito la Delibera n. 40/2014/R/gas, Disposizioni in materia di accertamenti della
Reg. 305/2011/UE: il nuovo sistema per la marcatura CE dei prodotti per le costruzioni in vigore dal 1 luglio 2013
 Reg. 305/2011/UE: il nuovo sistema per la marcatura CE dei prodotti per le costruzioni in vigore dal 1 luglio 2013 Ing. Davide Babbini www.artea.it OBIETTIVO Il Regolamento 305/2011 (CPR, art. 1), che
Reg. 305/2011/UE: il nuovo sistema per la marcatura CE dei prodotti per le costruzioni in vigore dal 1 luglio 2013 Ing. Davide Babbini www.artea.it OBIETTIVO Il Regolamento 305/2011 (CPR, art. 1), che
IL DIRETTORE GENERALE per il mercato, la concorrenza, il consumatore, la vigilanza e la normativa tecnica
 DECRETO 19 maggio 2010 Modifica degli allegati al decreto 22 gennaio 2008, n. 37, concernente il regolamento in materia di attività di installazione degli impianti all'interno degli edifici. IL DIRETTORE
DECRETO 19 maggio 2010 Modifica degli allegati al decreto 22 gennaio 2008, n. 37, concernente il regolamento in materia di attività di installazione degli impianti all'interno degli edifici. IL DIRETTORE
Allegato I. Parte A Obiettivi formativi
 Allegato I Parte A Obiettivi formativi Tenuto conto dei contenuti formativi riportati nell Allegato I del decreto legislativo n. 150/2012, si riportano di seguito i contenuti comuni degli specifici corsi
Allegato I Parte A Obiettivi formativi Tenuto conto dei contenuti formativi riportati nell Allegato I del decreto legislativo n. 150/2012, si riportano di seguito i contenuti comuni degli specifici corsi
2. Requisiti della formazione rivolta agli OSA e agli alimentaristi
 FORMAZIONE DEGLI ALIMENTARISTI E DEGLI OPERATORI DEL SETTORE ALIMENTARE (OSA) AI SENSI DELLA D.G.R. LIGURIA 29/06/2012 N. 793 1. Introduzione Un efficace formazione e un adeguato addestramento del personale
FORMAZIONE DEGLI ALIMENTARISTI E DEGLI OPERATORI DEL SETTORE ALIMENTARE (OSA) AI SENSI DELLA D.G.R. LIGURIA 29/06/2012 N. 793 1. Introduzione Un efficace formazione e un adeguato addestramento del personale
La valutazione del rischio chimico
 La valutazione del rischio chimico Introduzione Per sua stessa definizione, l agente chimico è una sostanza o un preparato di natura chimica. L agente chimico può presentarsi sotto forma di gas, vapore,
La valutazione del rischio chimico Introduzione Per sua stessa definizione, l agente chimico è una sostanza o un preparato di natura chimica. L agente chimico può presentarsi sotto forma di gas, vapore,
Esposizione ad agenti biologici
 Esposizione ad agenti biologici Il Titolo X corrisponde al Titolo VIII del D.Lgs. 626/94 di attuazione della direttiva 90/679/CEE, relativa alla protezione di lavoratori contro i rischi derivanti dall
Esposizione ad agenti biologici Il Titolo X corrisponde al Titolo VIII del D.Lgs. 626/94 di attuazione della direttiva 90/679/CEE, relativa alla protezione di lavoratori contro i rischi derivanti dall
Istituto Superiore Per la Prevenzione E la Sicurezza del Lavoro
 Istituto Superiore Per la Prevenzione E la Sicurezza del Lavoro Dipartimento Territoriale di BRESCIA Via San Francesco d Assisi, 11 25122 BRESCIA Competenze ed Obblighi degli Organismi Notificati, dei
Istituto Superiore Per la Prevenzione E la Sicurezza del Lavoro Dipartimento Territoriale di BRESCIA Via San Francesco d Assisi, 11 25122 BRESCIA Competenze ed Obblighi degli Organismi Notificati, dei
Circolare N.85 del 23 Maggio 2013
 Circolare N.85 del 23 Maggio 2013 Dal 01.06.2013 obbligatorie le procedure standardizzate per la valutazione dei rischi Gentile cliente con la presente intendiamo informarla che a partire dal 01.06.2013
Circolare N.85 del 23 Maggio 2013 Dal 01.06.2013 obbligatorie le procedure standardizzate per la valutazione dei rischi Gentile cliente con la presente intendiamo informarla che a partire dal 01.06.2013
Regolamento sui limiti al cumulo degli incarichi ricoperti dagli Amministratori del Gruppo Banco Popolare
 Regolamento sui limiti al cumulo degli incarichi ricoperti dagli Amministratori del Gruppo Banco Popolare febbraio 2013 1 1 PREMESSA... 3 1.1 Oggetto... 3 1.2 Perimetro di applicazione e modalità di recepimento...
Regolamento sui limiti al cumulo degli incarichi ricoperti dagli Amministratori del Gruppo Banco Popolare febbraio 2013 1 1 PREMESSA... 3 1.1 Oggetto... 3 1.2 Perimetro di applicazione e modalità di recepimento...
(Atti per i quali la pubblicazione non è una condizione di applicabilità) COMMISSIONE
 COMMISSIONE") L 86/6 Gazzetta ufficiale dell Unione europea 5.4.2005 II (Atti per i quali la pubblicazione non è una condizione di applicabilità) COMMISSIONE DECISIONE DELLA COMMISSIONE del 22 marzo 2005 che stabilisce
L 86/6 Gazzetta ufficiale dell Unione europea 5.4.2005 II (Atti per i quali la pubblicazione non è una condizione di applicabilità) COMMISSIONE DECISIONE DELLA COMMISSIONE del 22 marzo 2005 che stabilisce
Università degli Studi di Modena e Reggio Emilia
 RIFIUTI SPECIALI PRINCIPALE NORMATIVA DI RIFERIMENTO DECRETO LEGISLATIVO 3 APRILE 2006, N. 152. - Norme in materia ambientale e successive modifiche ed integrazioni CLASSIFICAZIONE DEI RIFIUTI SPECIALI
RIFIUTI SPECIALI PRINCIPALE NORMATIVA DI RIFERIMENTO DECRETO LEGISLATIVO 3 APRILE 2006, N. 152. - Norme in materia ambientale e successive modifiche ed integrazioni CLASSIFICAZIONE DEI RIFIUTI SPECIALI
La libera circolazione dei prodotti e le problematiche della sicurezza Dal Nuovo Approccio al New Legal Framework (NFL)
") La libera circolazione dei prodotti e le problematiche della sicurezza Dal Nuovo Approccio al New Legal Framework (NFL) 9 febbraio 2010 Ing. Fabio DATTILO Direttore Centrale della Direzione Centrale per
La libera circolazione dei prodotti e le problematiche della sicurezza Dal Nuovo Approccio al New Legal Framework (NFL) 9 febbraio 2010 Ing. Fabio DATTILO Direttore Centrale della Direzione Centrale per
Diciture o marche che consentono di identificare la partita alla quale appartiene una derrata alimentare ***I
 P7_TA-PROV(2011)0208 Diciture o marche che consentono di identificare la partita alla quale appartiene una derrata alimentare ***I Risoluzione legislativa del Parlamento europeo dell'11 maggio 2011 sulla
P7_TA-PROV(2011)0208 Diciture o marche che consentono di identificare la partita alla quale appartiene una derrata alimentare ***I Risoluzione legislativa del Parlamento europeo dell'11 maggio 2011 sulla